

Approval of Cancer Drugs With Uncertain Therapeutic Value: A Comparison of Regulatory Decisions in Europe and the United States
- PMID: 33021339
- PMCID: PMC7772660
- DOI: 10.1111/1468-0009.12476
Approval of Cancer Drugs With Uncertain Therapeutic Value: A Comparison of Regulatory Decisions in Europe and the United States
Abstract
Policy Points Regulatory agencies may have limited evidence on the clinical benefits and harms of new drugs when deciding whether new therapeutic agents are allowed to enter the market and under which conditions, including whether approval is granted under special regulatory pathways and obligations to address knowledge gaps through postmarketing studies are imposed. In a matched comparison of marketing applications for cancer drugs of uncertain therapeutic value reviewed by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), we found frequent discordance between the two agencies on regulatory outcomes and the use of special regulatory pathways. Both agencies often granted regular approval, even when the other agency judged there to be substantial uncertainty about drug benefits and risks that needed to be resolved through additional studies in the postmarketing period. Postmarketing studies imposed by regulators under special approval pathways to address remaining questions of efficacy and safety may not be suited to deliver timely, confirmatory evidence due to shortcomings in study design and delays, raising questions over the suitability of the FDA's Accelerated Approval and the EMA's Conditional Marketing Authorization as tools for allowing early market access for cancer drugs while maintaining rigorous regulatory standards.
Context: Regulatory agencies are increasingly required to make market approval decisions for new drugs on the basis of limited clinical evidence, a situation commonly encountered in cancer. We aimed to investigate how regulators manage uncertainty in the benefit-risk profiles of new cancer drugs by comparing decisions for the world's two largest regulatory bodies-the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA)-over a 5-year period.
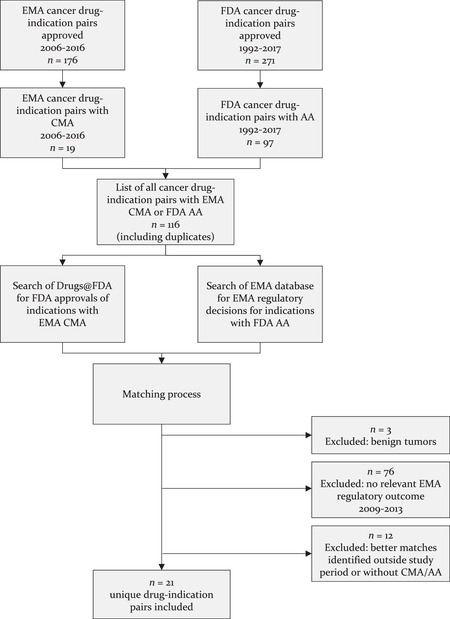
Methods: We systematically identified a set of cancer drug-indication pairs for which data on efficacy and safety was less complete than that required for regular approval at time of market entry from 2009 to 2013, as determined by the FDA's use of Accelerated Approval (AA) or the EMA's use of Conditional Marketing Authorization (CMA) pathways, and matched these across the two agencies. Using publicly available information, we compared regulatory pathways and outcomes, final approved indications, and postmarketing obligations imposed by the agencies.
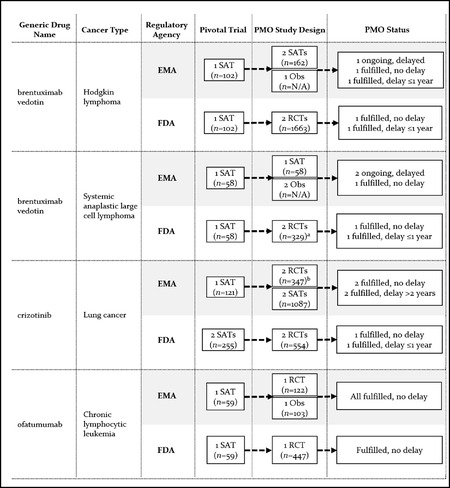
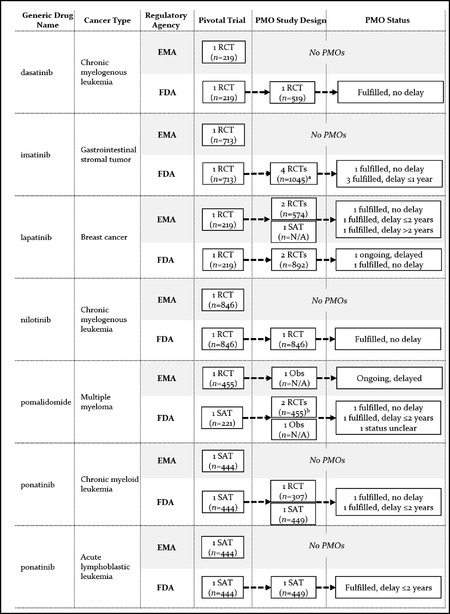
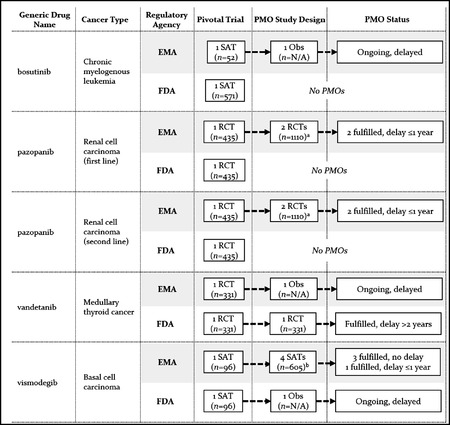
Findings: We identified 21 cancer drug-indication pairs that received FDA AA, EMA CMA, or both. Although most applications relied on identical pivotal trials across the FDA and the EMA, regulatory pathways often differed; 57% of indications received either FDA AA or EMA CMA, and regular approval by the other agency. After approval, the EMA more often accepted single-arm studies to confirm clinical benefit compared to the FDA (75% vs. 29% of indications), and the FDA more commonly requested randomized controlled trials (85% vs. 50%). Forty-one percent of confirmatory trials after FDA AA were conducted in different populations than the approved indication, compared to 13% after EMA CMA. Both agencies relied primarily on surrogate measures of patient benefit for postmarketing obligations. After a median follow-up of 7.25 years, 40% of FDA and 61% of EMA postmarketing obligations after AA and CMA, respectively, were delayed.
Conclusions: US and European regulators often deemed early and less complete evidence on benefit-risk profiles of cancer drugs sufficient to grant regular approval, raising questions over regulatory standards for the approval of new medicines. Even when imposing confirmatory studies in the postmarketing period through special approval pathways, meaningful evidence may not materialize due to shortcomings in study design and delays in conducting required studies with due diligence.
Keywords: European Medicines Agency; US Food and Drug Administration; cancer; pharmaceutical regulation.
© 2020 The Authors. The Milbank Quarterly published by Wiley Periodicals LLC on behalf of The Millbank Memorial Fund.
Figures
Comment in
-
In the December 2020 Issue of the Quarterly.Milbank Q. 2020 Dec;98(4):1027-1032. doi: 10.1111/1468-0009.12487. Milbank Q. 2020. PMID: 33377289 Free PMC article. No abstract available.
References
-
- European Medicines Agency . PRIME: A Two‐Year Overview . London, England: European Medicines Agency; 2018. https://www.ema.europa.eu/en/documents/report/prime-two-year-overview_en.... Accessed October 8, 2019.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
