

Atypical teratoid rhabdoid tumor: molecular insights and translation to novel therapeutics
- PMID: 33021733
- PMCID: PMC8230985
- DOI: 10.1007/s11060-020-03639-w
Atypical teratoid rhabdoid tumor: molecular insights and translation to novel therapeutics
Abstract
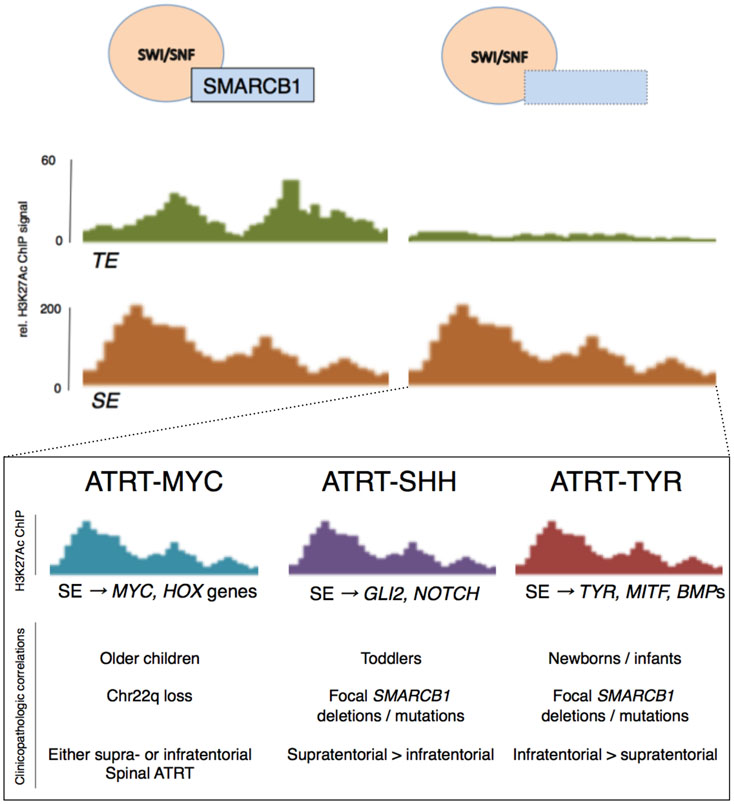
Introduction: Atypical teratoid rhabdoid tumor (ATRT) is a rare, often lethal brain tumor of childhood characterized by a complex epigenetic landscape amongst a simple genetic background. Recent molecular studies have defined key biologic events that contribute to tumorigenesis and molecular subtypes of ATRT.
Methods: Seminal studies on ATRT are reviewed with an emphasis on molecular pathogenesis and its relevance to novel therapeutics.
Results: In this review, we summarize the key clinicopathologic and molecular features of ATRT, completed and ongoing clinical trials and outline the translational potential of novel insights into the molecular pathogenesis of this tumor.
Conclusions: SMARCB1 loss is the key genetic event in ATRT pathogenesis that leads to widespread epigenetic dysregulation and loss of lineage-specific enhancers. Current work is defining subtype-specific treatments that target underlying molecular derangements that drive tumorigenesis.
Keywords: ATRT; Atypical teratoid rhabdoid tumor; Chromatin remodeling; Epigenetics; SWI/SNF complex.
Conflict of interest statement
Figures
References
-
- Ellison D, Love S (2013) Neuropathology: A Reference Text of CNS Pathology, 3rd ed. Mosby Ltd
-
- Biegel JA, Tan L, Zhang F, et al. (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467 - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
