

Single-Cell Study of Two Rat Models of Pulmonary Arterial Hypertension Reveals Connections to Human Pathobiology and Drug Repositioning
- PMID: 33021809
- PMCID: PMC8048757
- DOI: 10.1164/rccm.202006-2169OC
Single-Cell Study of Two Rat Models of Pulmonary Arterial Hypertension Reveals Connections to Human Pathobiology and Drug Repositioning
Abstract
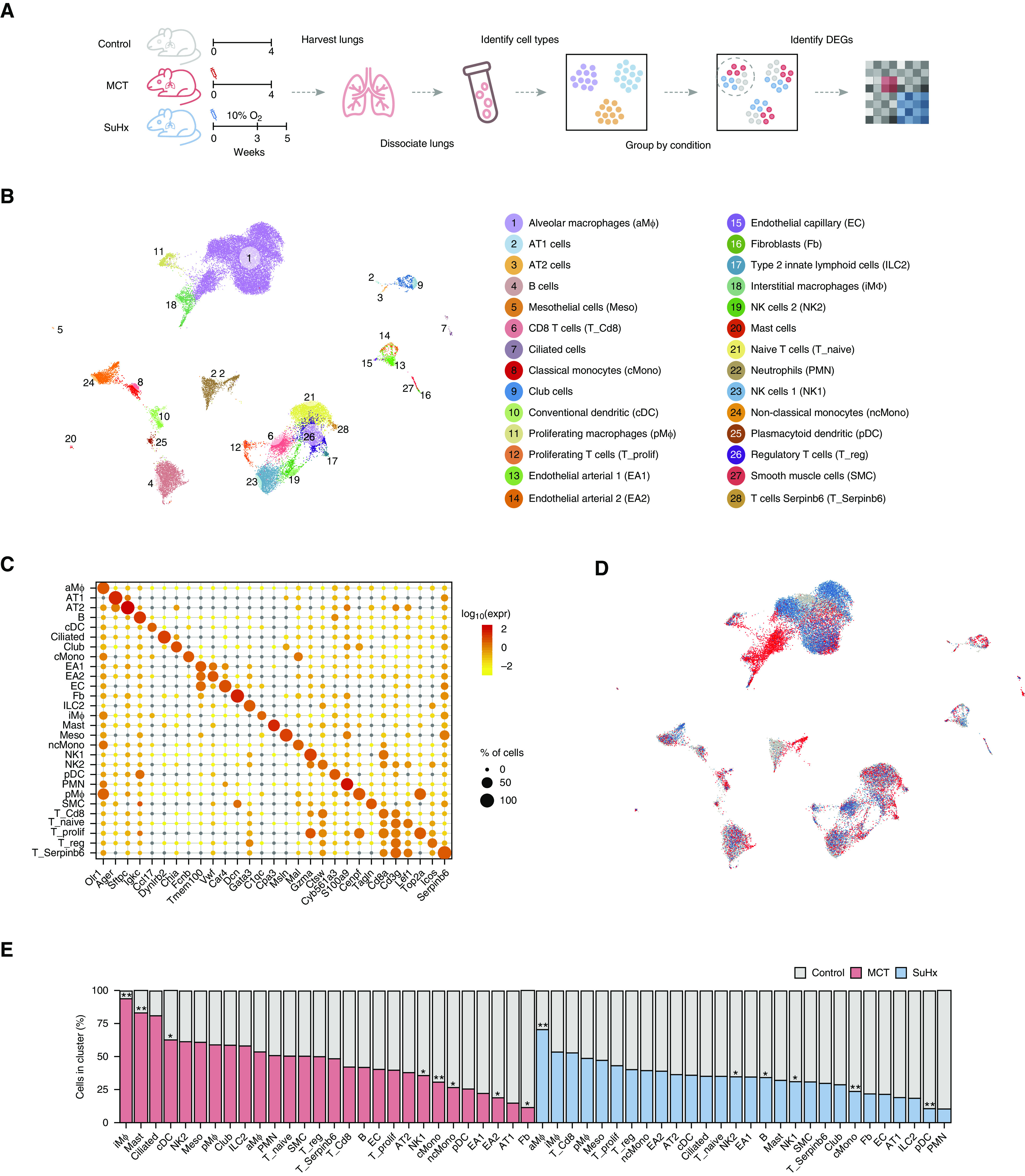
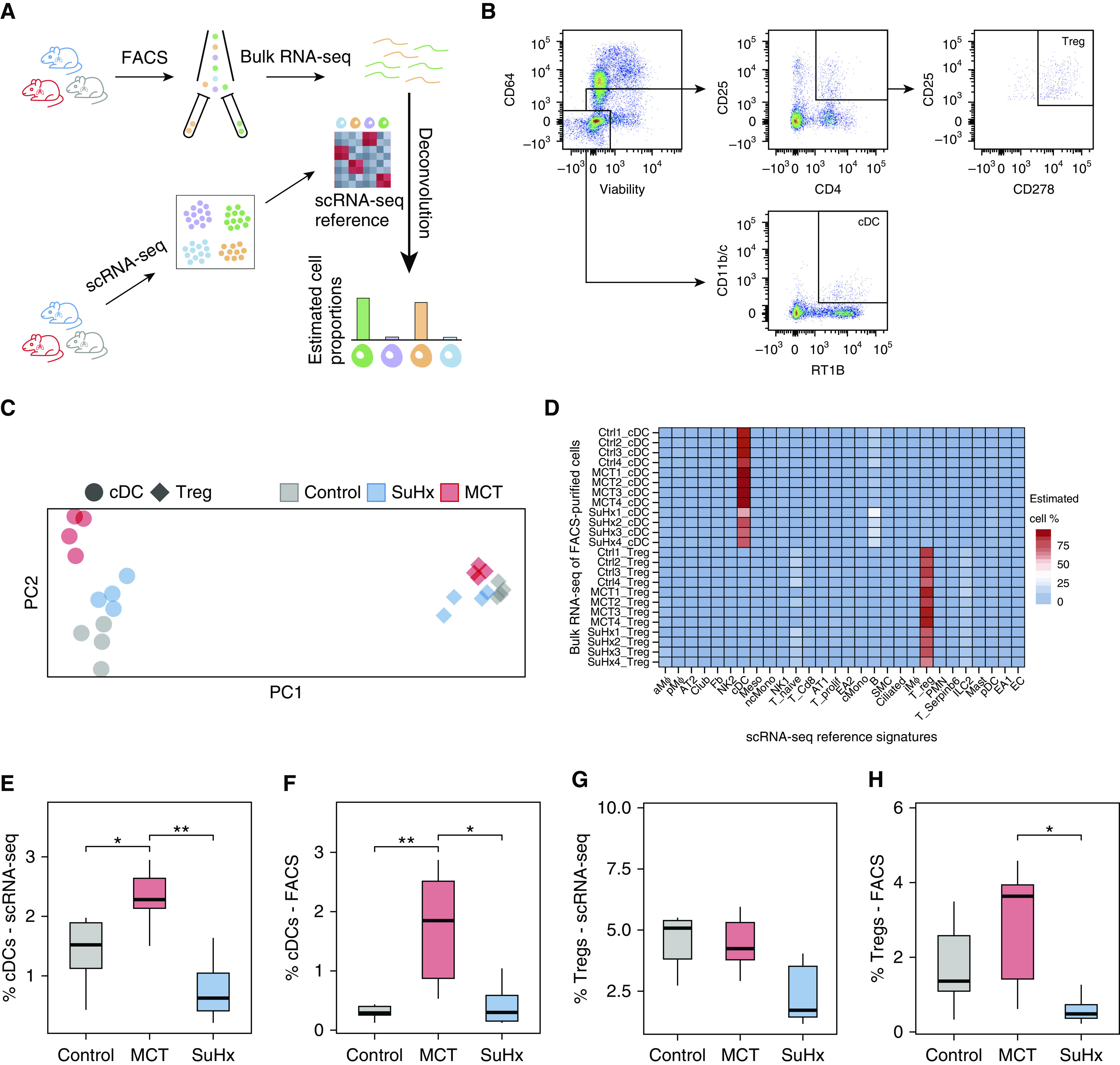
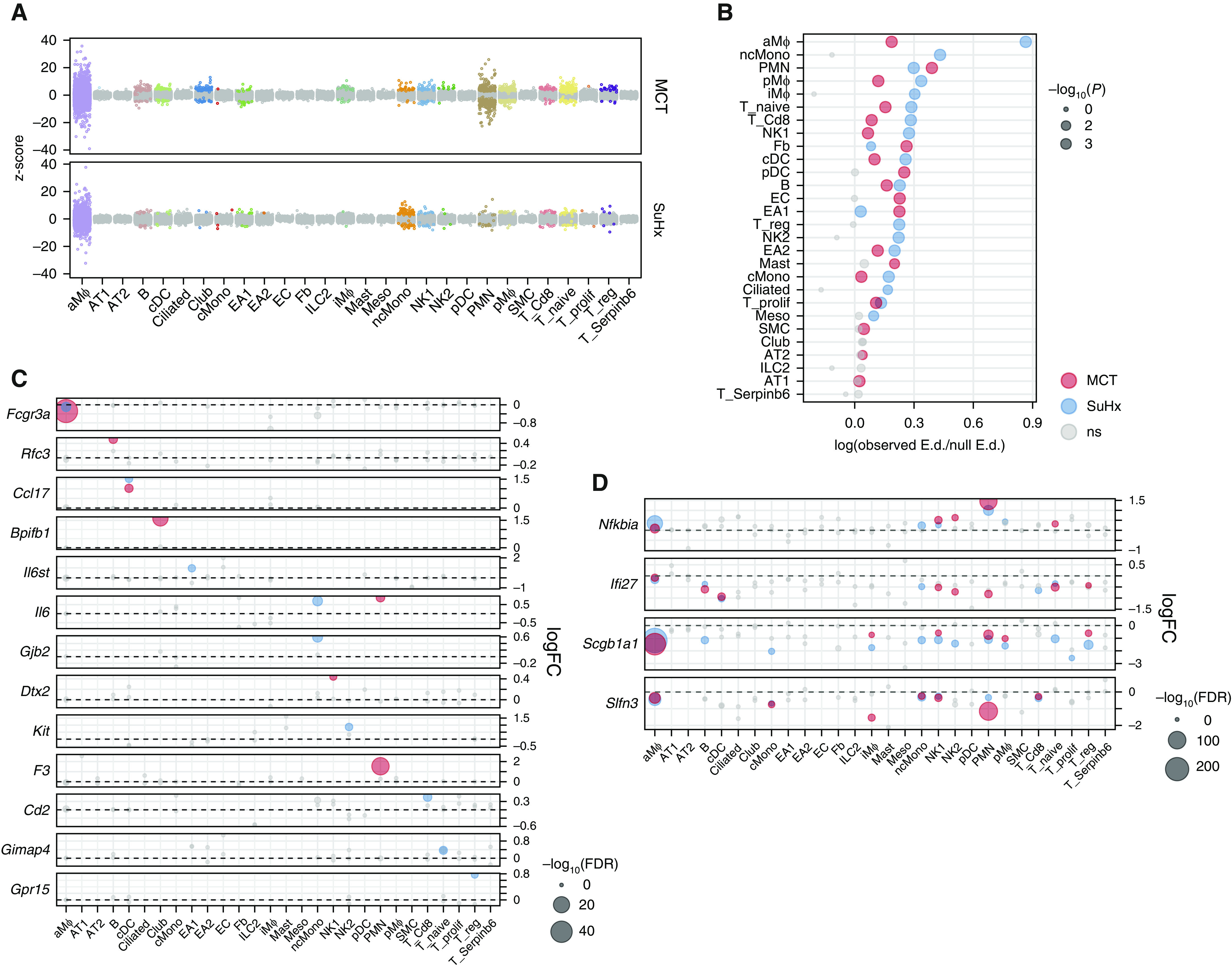
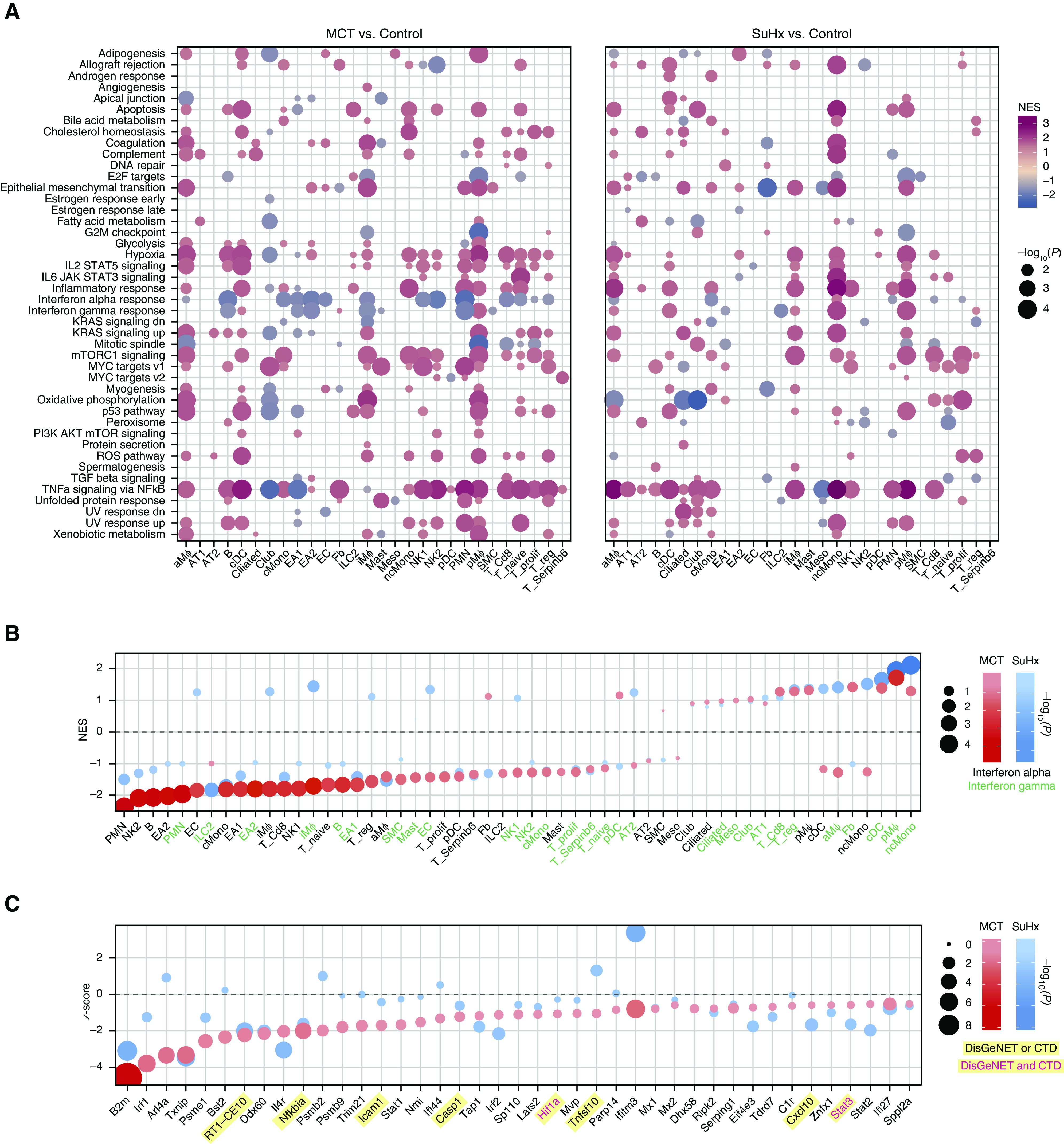
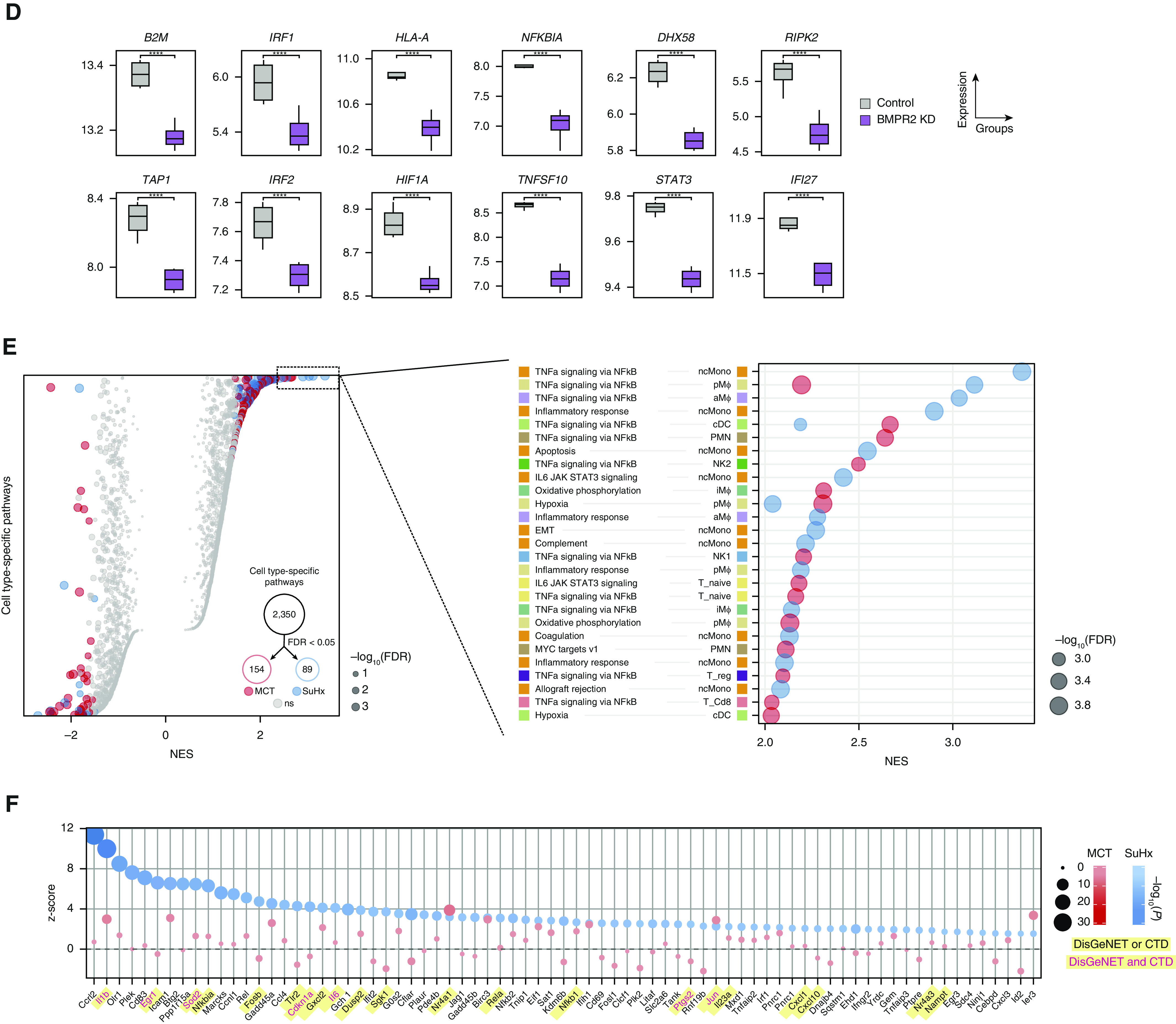
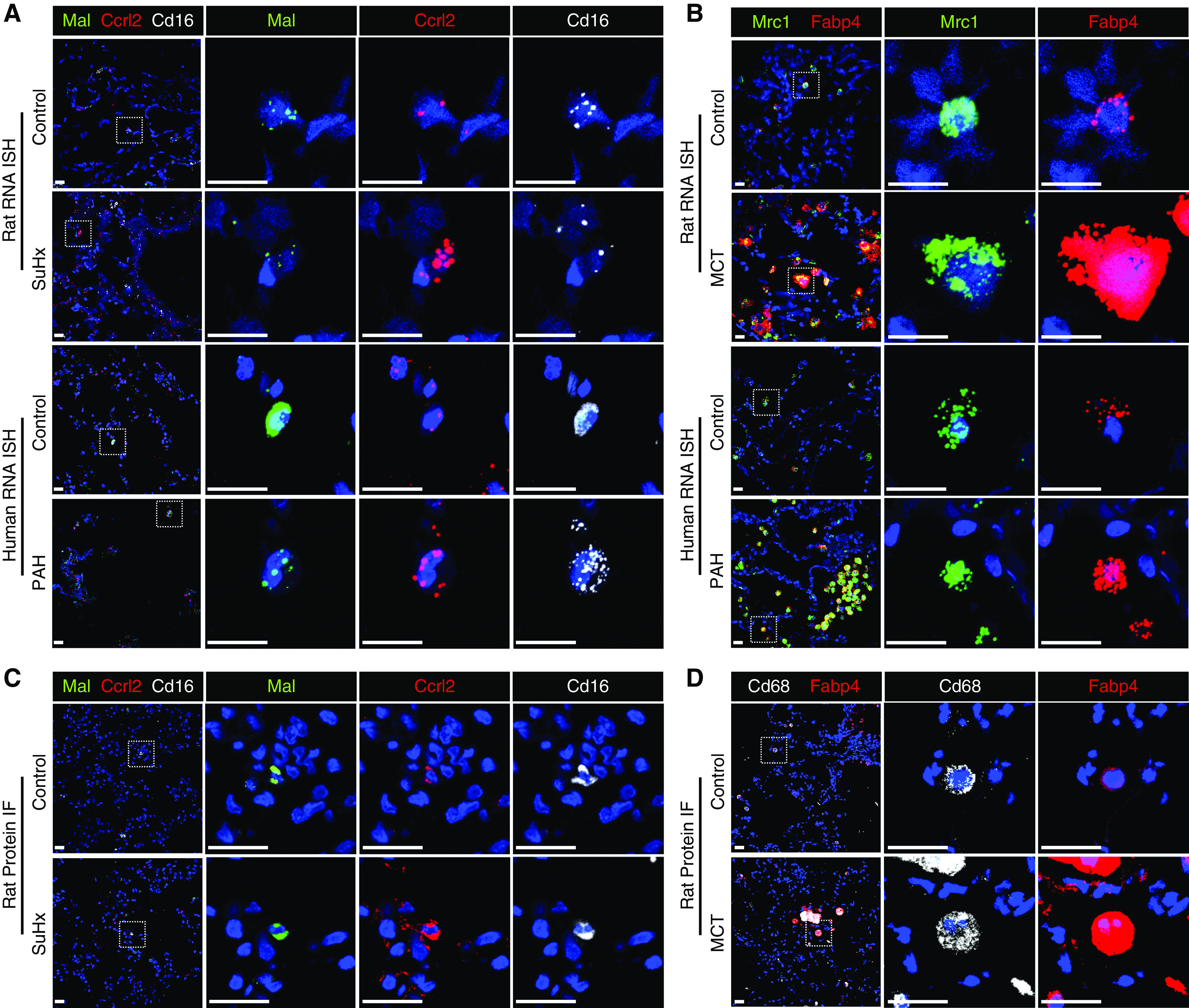
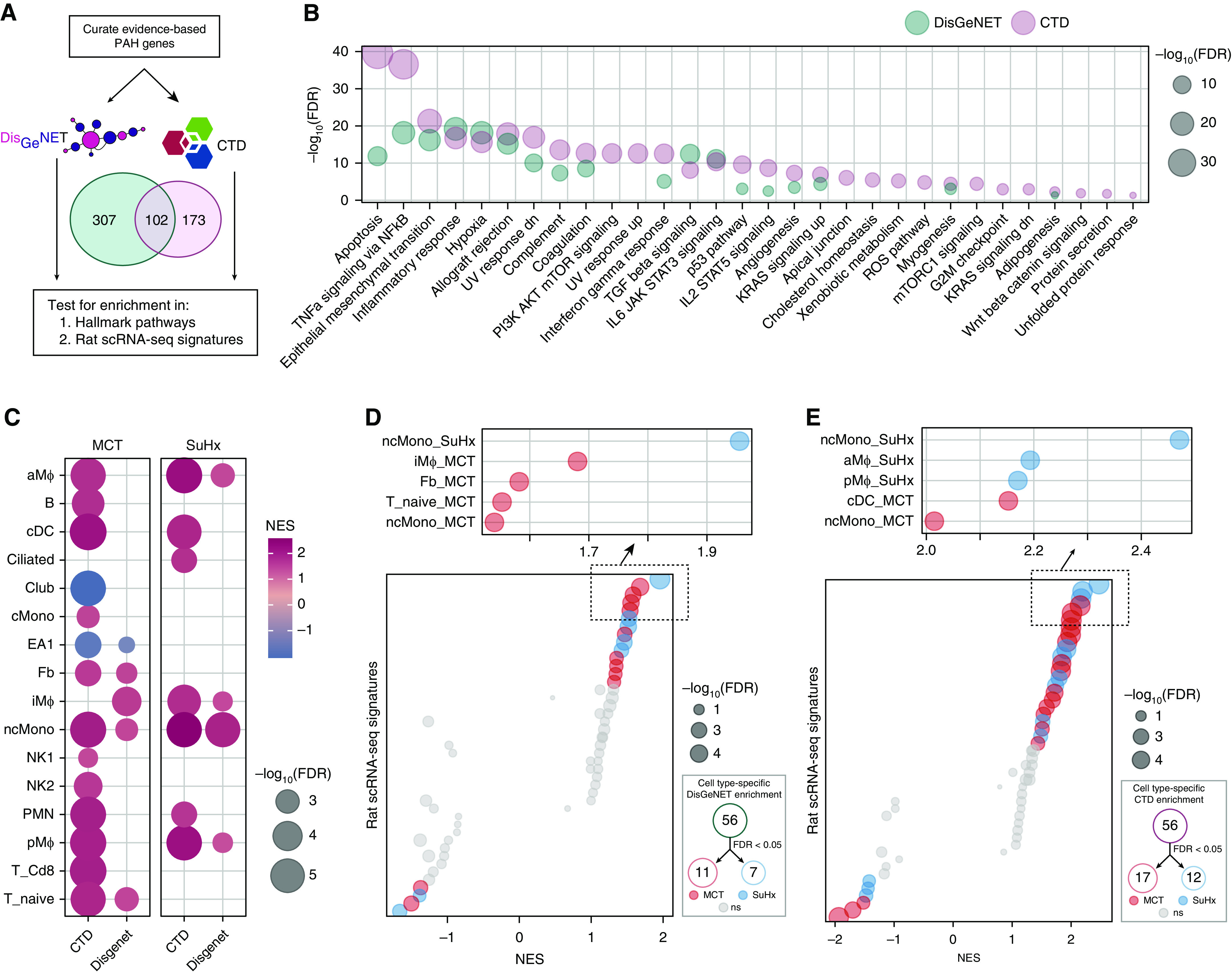
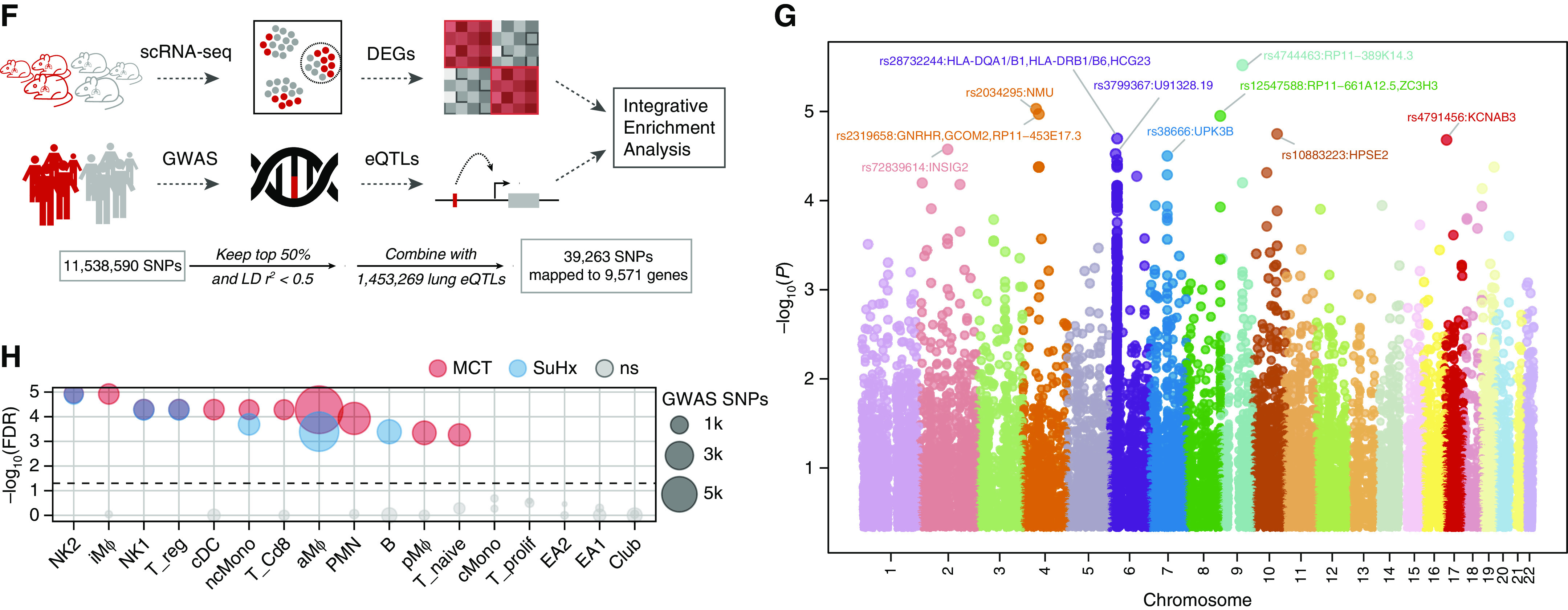
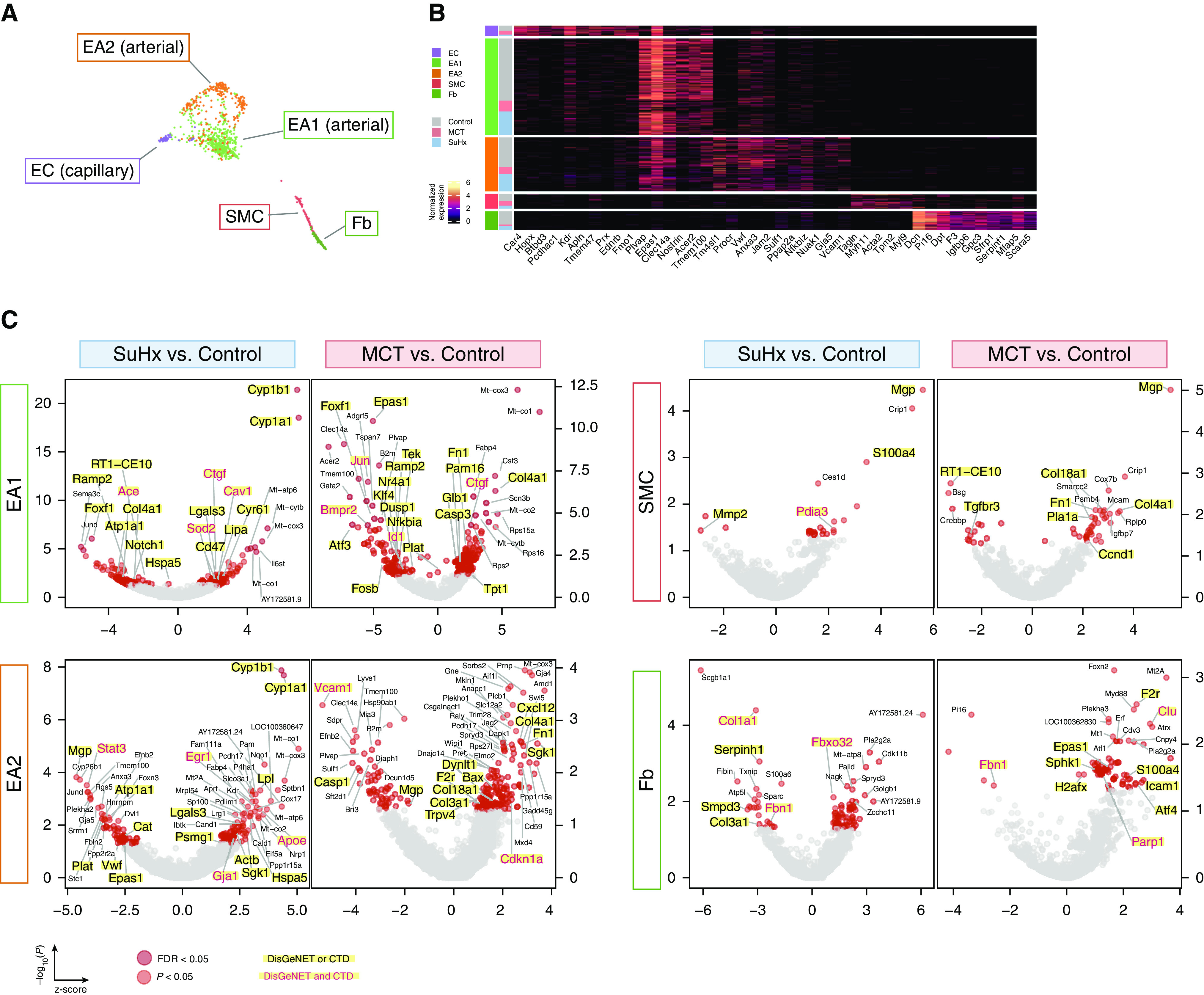
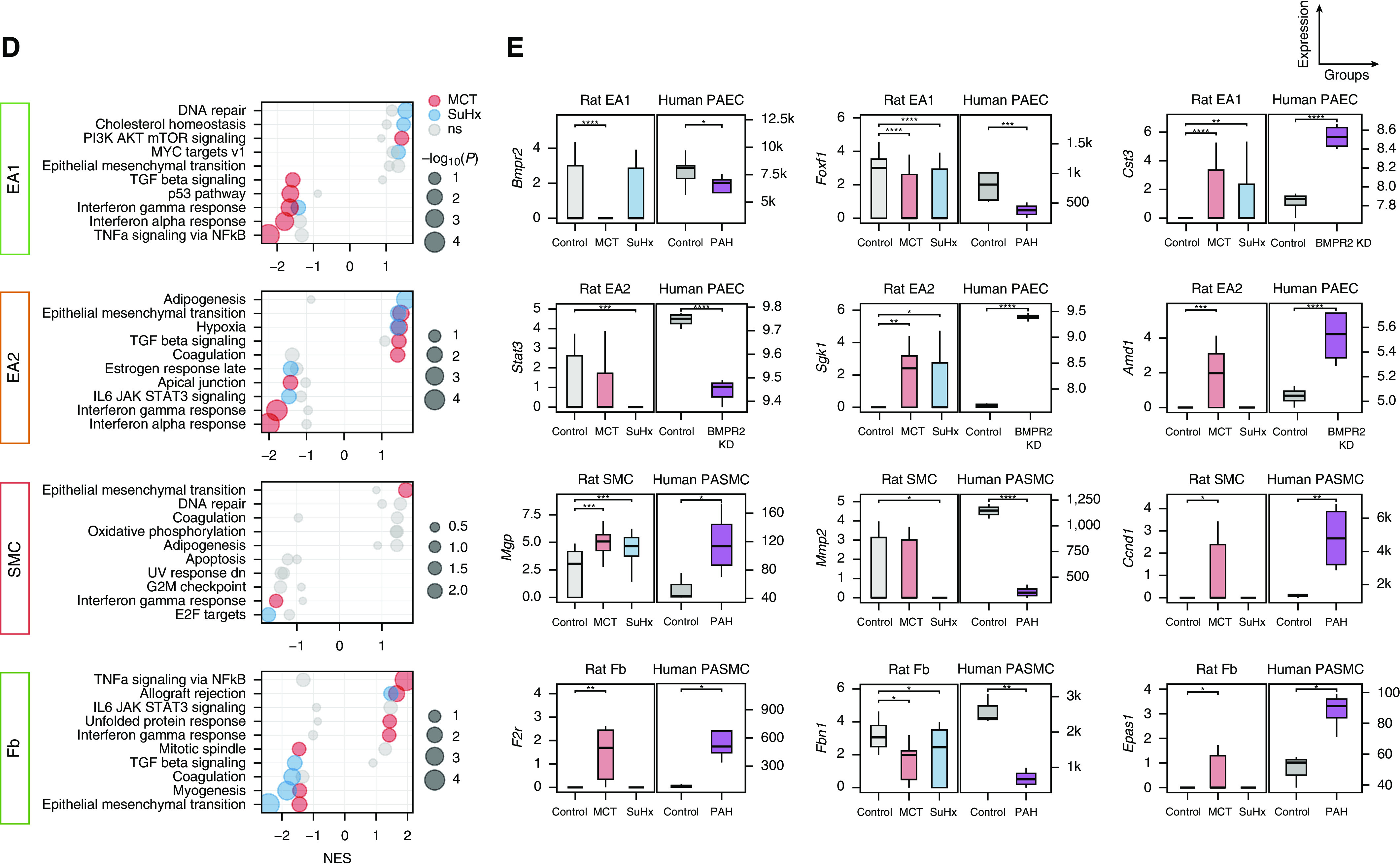
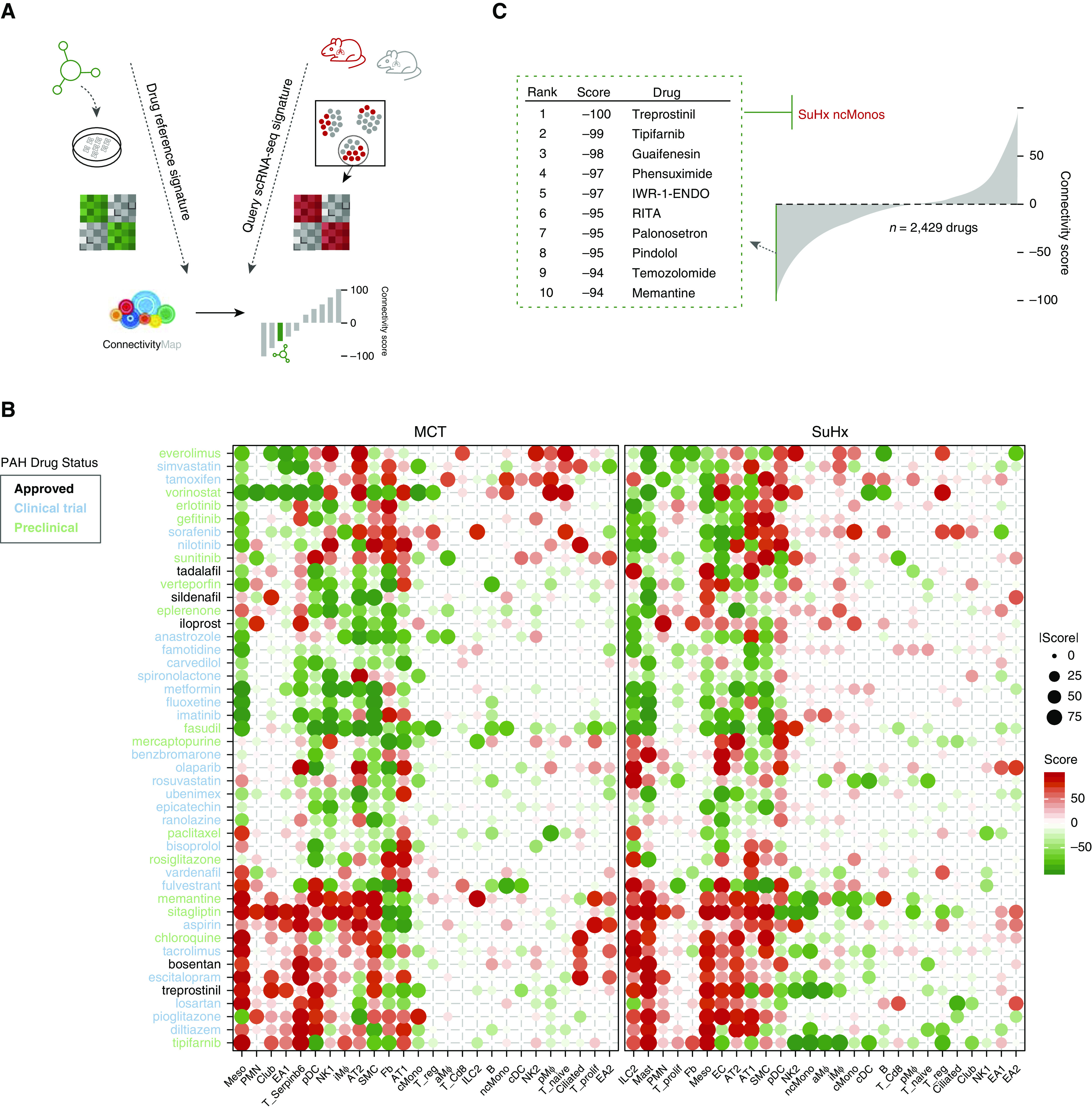
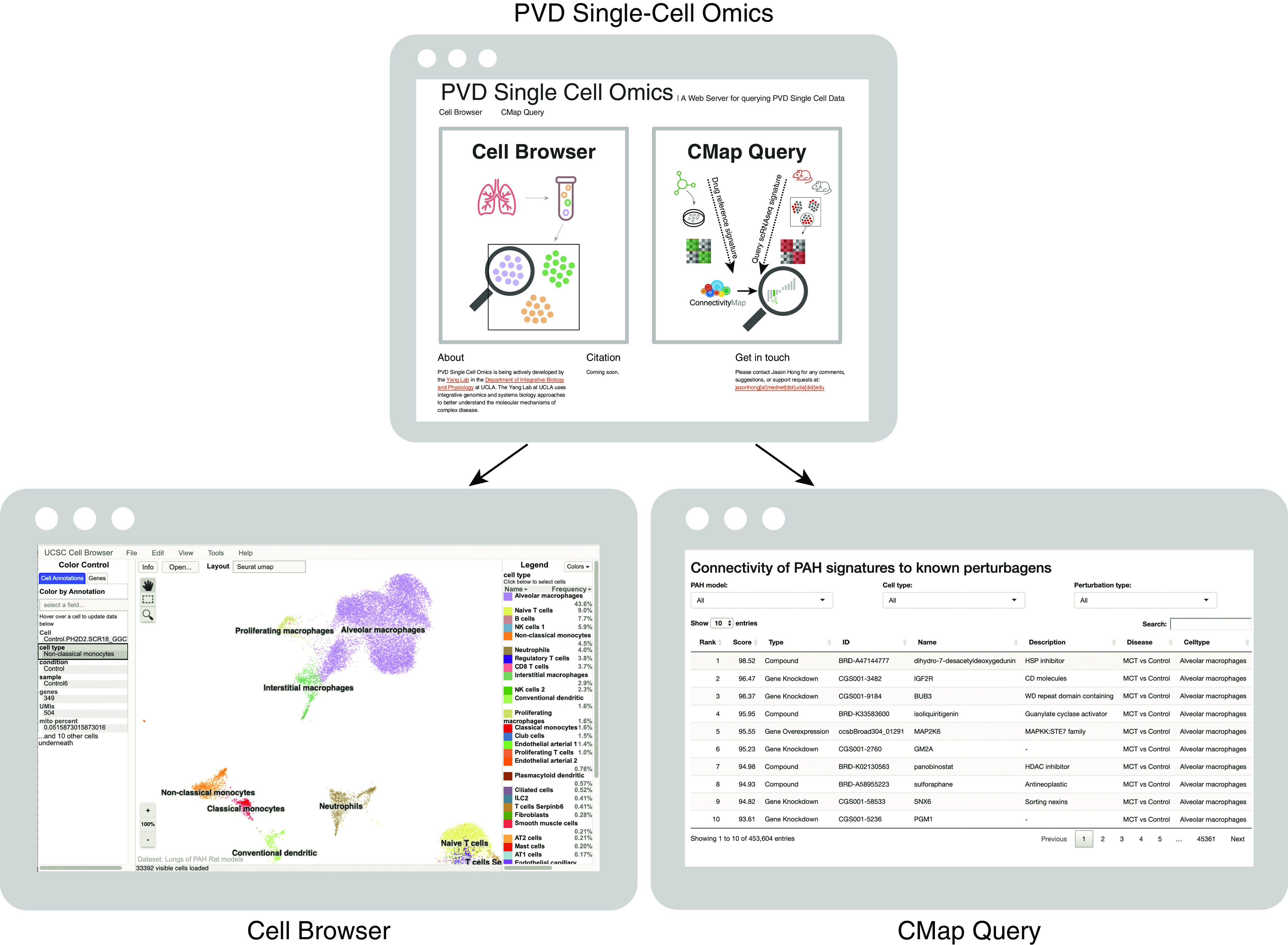
Rationale: The cellular and molecular landscape and translational value of commonly used models of pulmonary arterial hypertension (PAH) are poorly understood. Single-cell transcriptomics can enhance molecular understanding of preclinical models and facilitate their rational use and interpretation.Objectives: To determine and prioritize dysregulated genes, pathways, and cell types in lungs of PAH rat models to assess relevance to human PAH and identify drug repositioning candidates.Methods: Single-cell RNA sequencing was performed on the lungs of monocrotaline (MCT), Sugen-hypoxia (SuHx), and control rats to identify altered genes and cell types, followed by validation using flow-sorted cells, RNA in situ hybridization, and immunofluorescence. Relevance to human PAH was assessed by histology of lungs from patients and via integration with human PAH genetic loci and known disease genes. Candidate drugs were predicted using Connectivity Map.Measurements and Main Results: Distinct changes in genes and pathways in numerous cell types were identified in SuHx and MCT lungs. Widespread upregulation of NF-κB signaling and downregulation of IFN signaling was observed across cell types. SuHx nonclassical monocytes and MCT conventional dendritic cells showed particularly strong NF-κB pathway activation. Genes altered in SuHx nonclassical monocytes were significantly enriched for PAH-associated genes and genetic variants, and candidate drugs predicted to reverse the changes were identified. An open-access online platform was developed to share single-cell data and drug candidates (http://mergeomics.research.idre.ucla.edu/PVDSingleCell/).Conclusions: Our study revealed the distinct and shared dysregulation of genes and pathways in two commonly used PAH models for the first time at single-cell resolution and demonstrated their relevance to human PAH and utility for drug repositioning.
Keywords: Sugen-hypoxia; drug repurposing; monocrotaline; pulmonary hypertension; single-cell RNA sequencing.
Figures
Comment in
-
Zooming into Cellular and Molecular Heterogeneity of Pulmonary Hypertension. What More Single-Cell Omics Can Offer.Am J Respir Crit Care Med. 2021 Apr 15;203(8):941-943. doi: 10.1164/rccm.202010-3889ED. Am J Respir Crit Care Med. 2021. PMID: 33171066 Free PMC article. No abstract available.
References
-
- Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest. 2012;142:448–456. - PubMed
-
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277–1289. - PubMed
-
- Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. 2019;25:1153–1163. - PubMed
