

Defective mitophagy in Alzheimer's disease
- PMID: 33022416
- PMCID: PMC7710581
- DOI: 10.1016/j.arr.2020.101191
Defective mitophagy in Alzheimer's disease
Abstract
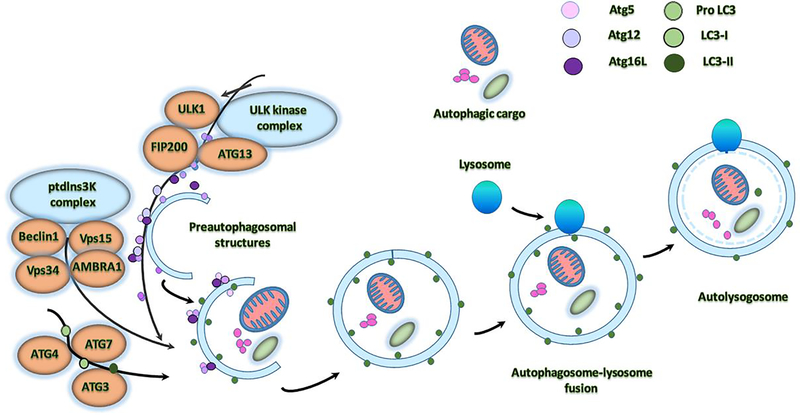
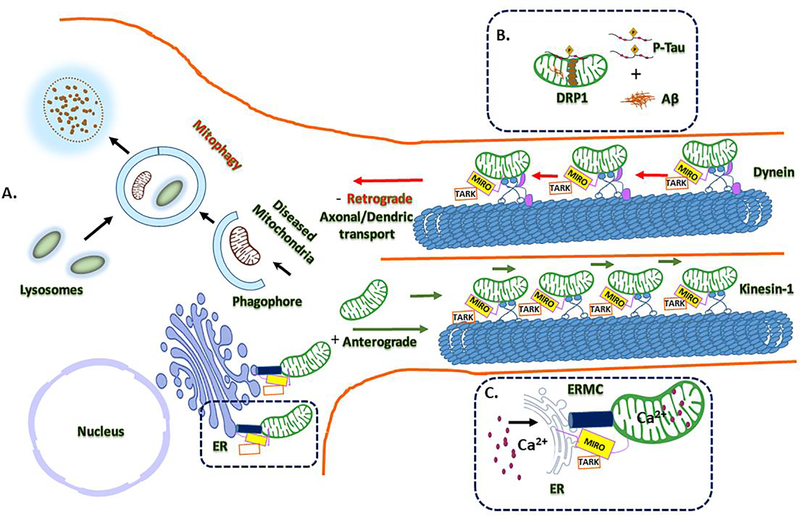
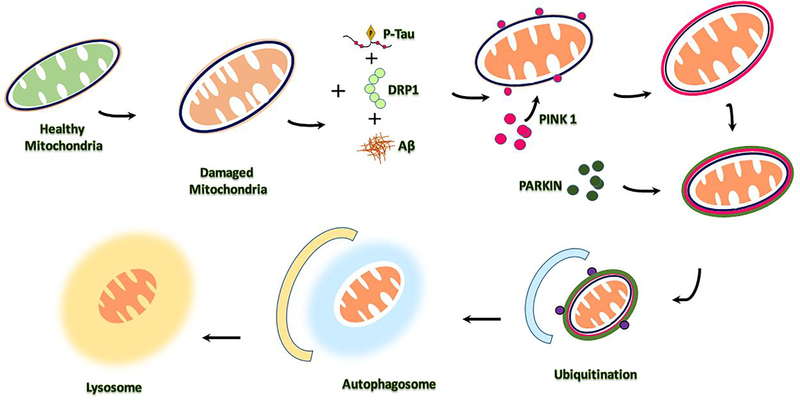
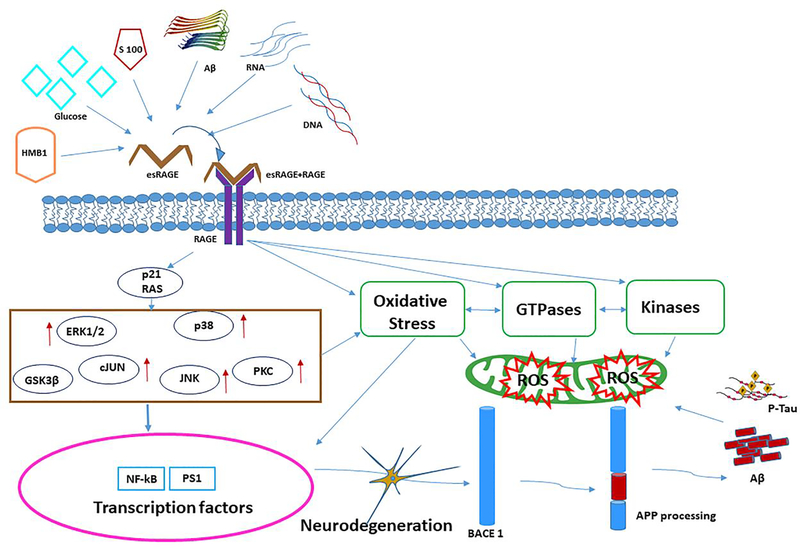
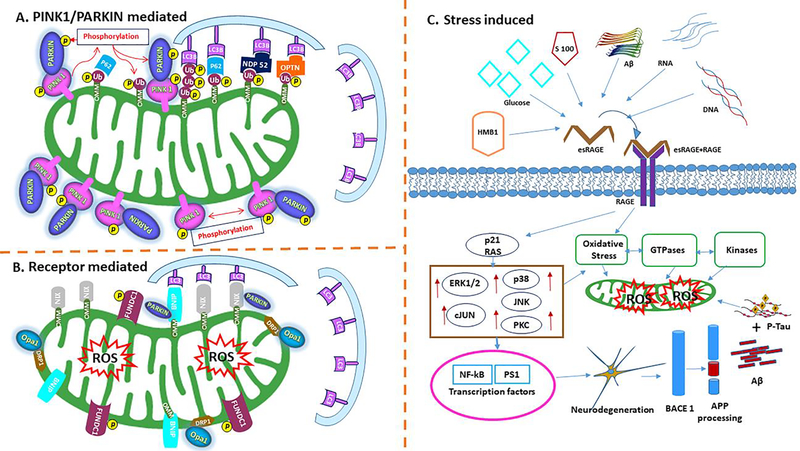
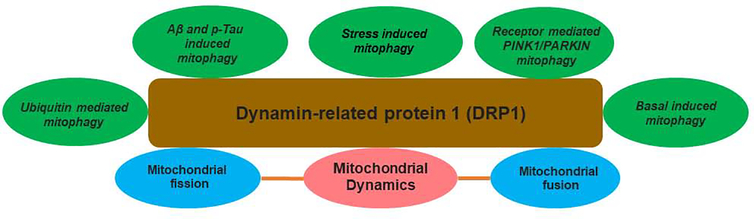
Alzheimer's disease (AD) is a progressive, mental illness without cure. Several years of intense research on postmortem AD brains, cell and mouse models of AD have revealed that multiple cellular changes are involved in the disease process, including mitochondrial abnormalities, synaptic damage, and glial/astrocytic activation, in addition to age-dependent accumulation of amyloid beta (Aβ) and hyperphosphorylated tau (p-tau). Synaptic damage and mitochondrial dysfunction are early cellular changes in the disease process. Healthy and functionally active mitochondria are essential for cellular functioning. Dysfunctional mitochondria play a central role in aging and AD. Mitophagy is a cellular process whereby damaged mitochondria are selectively removed from cell and mitochondrial quality and biogenesis. Mitophagy impairments cause the progressive accumulation of defective organelle and damaged mitochondria in cells. In AD, increased levels of Aβ and p-tau can induce reactive oxygen species (ROS) production, causing excessive fragmentation of mitochondria and promoting defective mitophagy. The current article discusses the latest developments of mitochondrial research and also highlights multiple types of mitophagy, including Aβ and p-tau-induced mitophagy, stress-induced mitophagy, receptor-mediated mitophagy, ubiquitin mediated mitophagy and basal mitophagy. This article also discusses the physiological states of mitochondria, including fission-fusion balance, Ca2+ transport, and mitochondrial transport in normal and diseased conditions. Our article summarizes current therapeutic interventions, like chemical or natural mitophagy enhancers, that influence mitophagy in AD. Our article discusses whether a partial reduction of Drp1 can be a mitophagy enhancer and a therapeutic target for mitophagy in AD and other neurological diseases.
Keywords: Alzheimer’s disease; Amyloid beta; Mitochondrial dysfunction; Mitophagy; Phosphorylated tau.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
Figures
References
-
- Angelika BH, Hees JT 2019. Calcium Dysregulation and Mitochondrial Dysfunction Form A Vicious Cycle in Parkinson’s Disease. American Journal of Biomedical Science & Research 5, 246–249.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
