

Retinal Degeneration and Alzheimer's Disease: An Evolving Link
- PMID: 33023198
- PMCID: PMC7582766
- DOI: 10.3390/ijms21197290
Retinal Degeneration and Alzheimer's Disease: An Evolving Link
Abstract
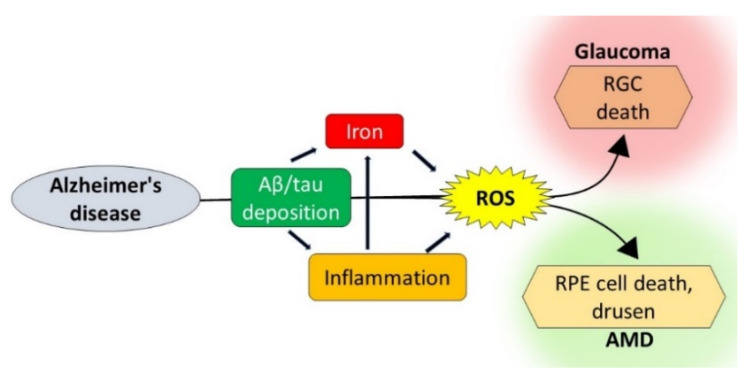
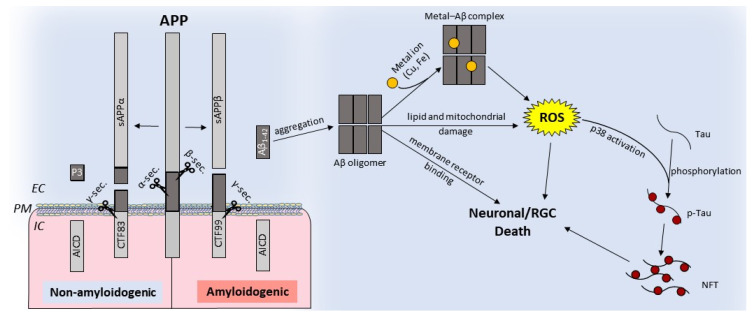
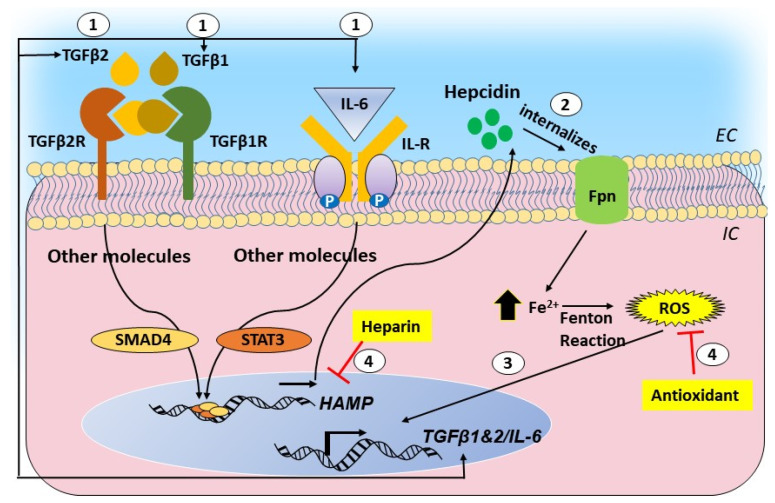
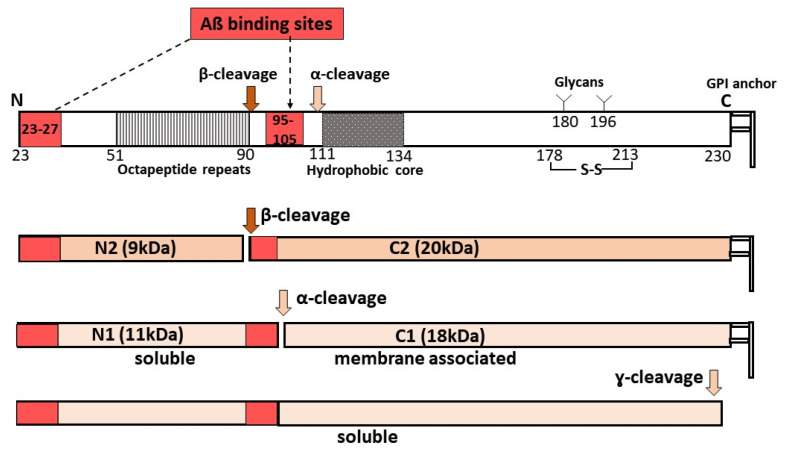
Age-related macular degeneration (AMD) and glaucoma are degenerative conditions of the retina and a significant cause of irreversible blindness in developed countries. Alzheimer's disease (AD), the most common dementia of the elderly, is often associated with AMD and glaucoma. The cardinal features of AD include extracellular accumulation of amyloid β (Aβ) and intracellular deposits of hyper-phosphorylated tau (p-tau). Neuroinflammation and brain iron dyshomeostasis accompany Aβ and p-tau deposits and, together, lead to progressive neuronal death and dementia. The accumulation of Aβ and iron in drusen, the hallmark of AMD, and Aβ and p-tau in retinal ganglion cells (RGC), the main retinal cell type implicated in glaucoma, and accompanying inflammation suggest overlapping pathology. Visual abnormalities are prominent in AD and are believed to develop before cognitive decline. Some are caused by degeneration of the visual cortex, while others are due to RGC loss or AMD-associated retinal degeneration. Here, we review recent information on Aβ, p-tau, chronic inflammation, and iron dyshomeostasis as common pathogenic mechanisms linking the three degenerative conditions, and iron chelation as a common therapeutic option for these disorders. Additionally discussed is the role of prion protein, infamous for prion disorders, in Aβ-mediated toxicity and, paradoxically, in neuroprotection.
Keywords: Alzheimer’s disease; age related macular degeneration; drusen; glaucoma; inflammation; iron; oxidative stress; prion protein; reactive oxygen species; retinal degeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
