

Cholesterol-Induced Phenotypic Modulation of Smooth Muscle Cells to Macrophage/Fibroblast-like Cells Is Driven by an Unfolded Protein Response
- PMID: 33028096
- PMCID: PMC7752246
- DOI: 10.1161/ATVBAHA.120.315164
Cholesterol-Induced Phenotypic Modulation of Smooth Muscle Cells to Macrophage/Fibroblast-like Cells Is Driven by an Unfolded Protein Response
Abstract
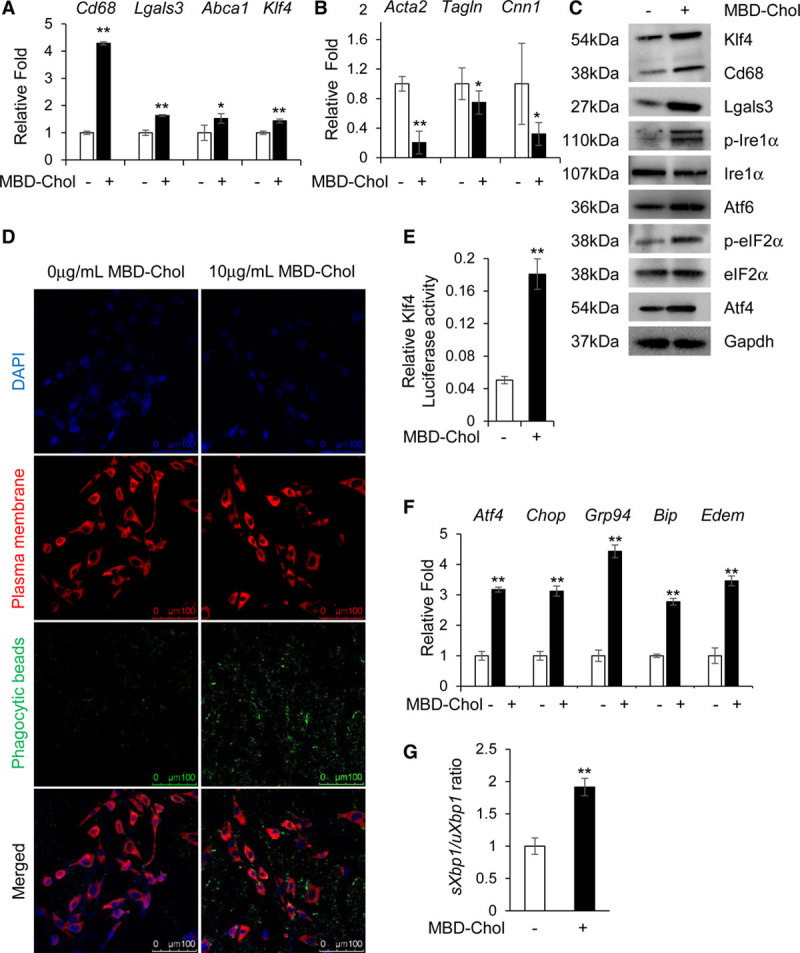
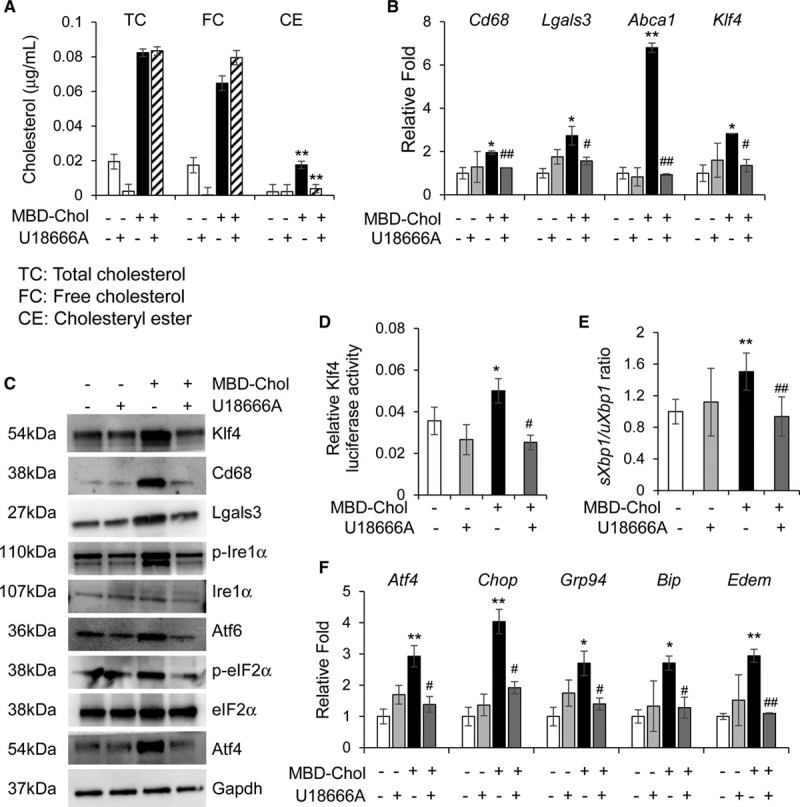
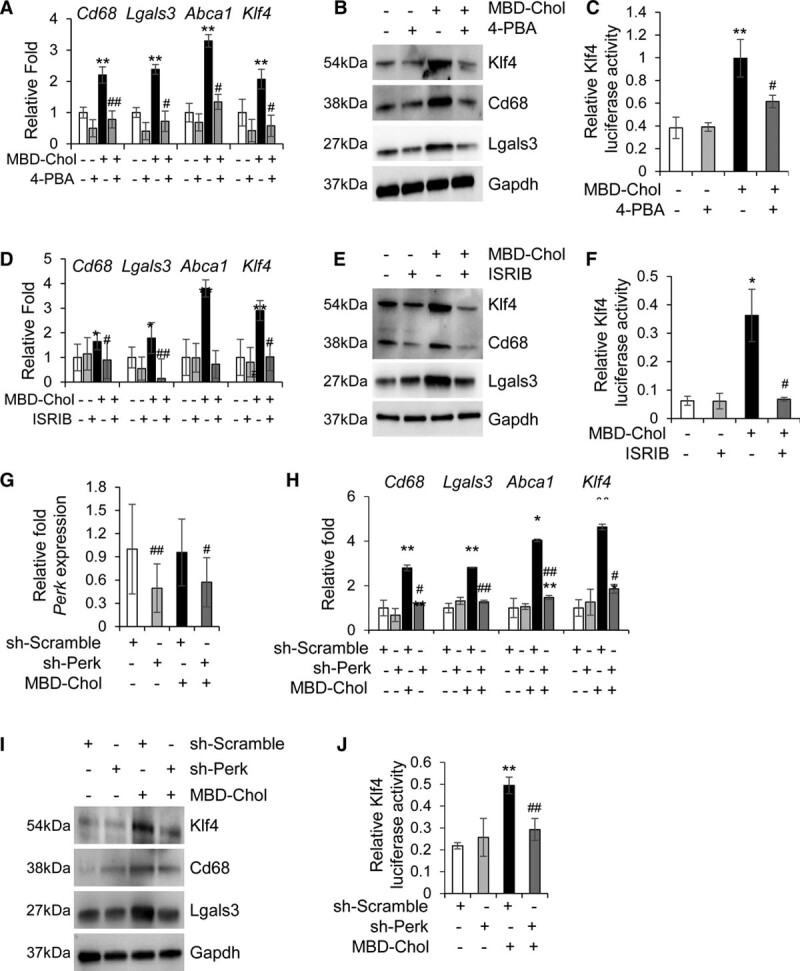
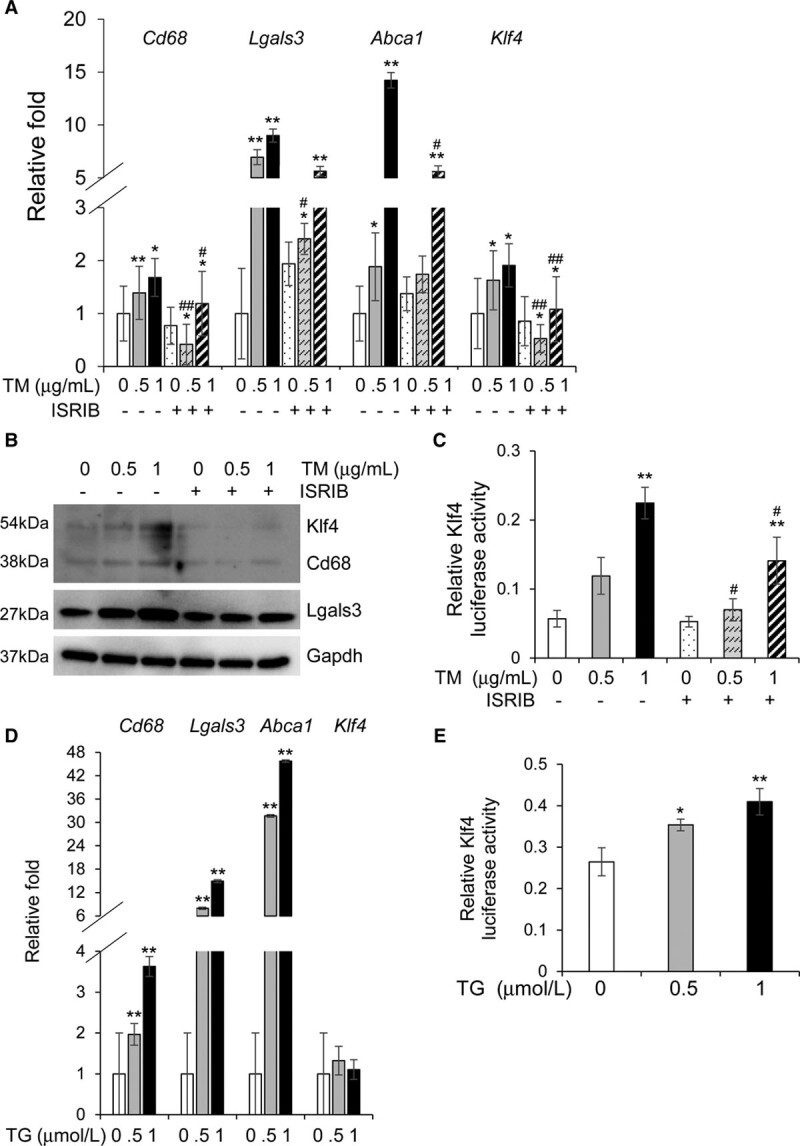
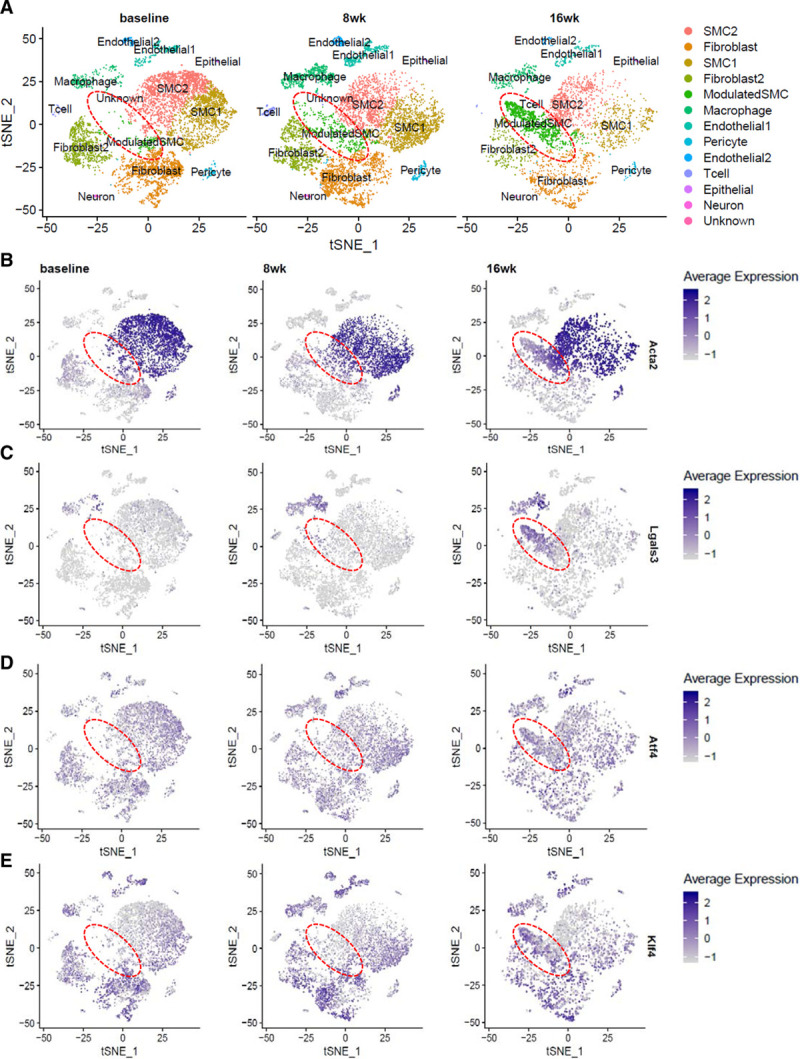
Objective: Vascular smooth muscle cells (SMCs) dedifferentiate and initiate expression of macrophage markers with cholesterol exposure. This phenotypic switching is dependent on the transcription factor Klf4 (Krüppel-like factor 4). We investigated the molecular pathway by which cholesterol induces SMC phenotypic switching. Approach and Results: With exposure to free cholesterol, SMCs decrease expression of contractile markers, activate Klf4, and upregulate a subset of macrophage and fibroblast markers characteristic of modulated SMCs that appear with atherosclerotic plaque formation. These phenotypic changes are associated with activation of all 3 pathways of the endoplasmic reticulum unfolded protein response (UPR), Perk (protein kinase RNA-like endoplasmic reticulum kinase), Ire (inositol-requiring enzyme) 1α, and Atf (activating transcription factor) 6. Blocking the movement of cholesterol from the plasma membrane to the endoplasmic reticulum prevents free cholesterol-induced UPR, Klf4 activation, and upregulation of the majority of macrophage and fibroblast markers. Cholesterol-induced phenotypic switching is also prevented by global UPR inhibition or specific inhibition of Perk signaling. Exposure to chemical UPR inducers, tunicamycin and thapsigargin, is sufficient to induce these same phenotypic transitions. Finally, analysis of published single-cell RNA sequencing data during atherosclerotic plaque formation in hyperlipidemic mice provides preliminary in vivo evidence of a role of UPR activation in modulated SMCs.
Conclusions: Our data demonstrate that UPR is necessary and sufficient to drive phenotypic switching of SMCs to cells that resemble modulated SMCs found in atherosclerotic plaques. Preventing a UPR in hyperlipidemic mice diminishes atherosclerotic burden, and our data suggest that preventing SMC transition to dedifferentiated cells expressing macrophage and fibroblast markers contributes to this decreased plaque burden.
Keywords: atherosclerosis; cholesterol; fibroblast; macrophage; phenotype; smooth muscle cell.
Conflict of interest statement
None.
Figures

References
-
- Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem. 1984;259:14383–14388 - PubMed
-
- Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K. Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol. 2007;27:1159–1165. doi: 10.1161/ATVBAHA.106.134080 - PubMed
-
- Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res. 2008;79:14–23. doi: 10.1093/cvr/cvn099 - PubMed
-
- Dubland JA, Francis GA. So Much Cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol. 2016;27:155–161. doi: 10.1097/MOL.0000000000000279 - PubMed
-
- Ball RY, Stowers EC, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 1995;114:45–54. doi: 10.1016/0021-9150(94)05463-s - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
