

Emerging role of tumor cell plasticity in modifying therapeutic response
- PMID: 33028808
- PMCID: PMC7541492
- DOI: 10.1038/s41392-020-00313-5
Emerging role of tumor cell plasticity in modifying therapeutic response
Abstract
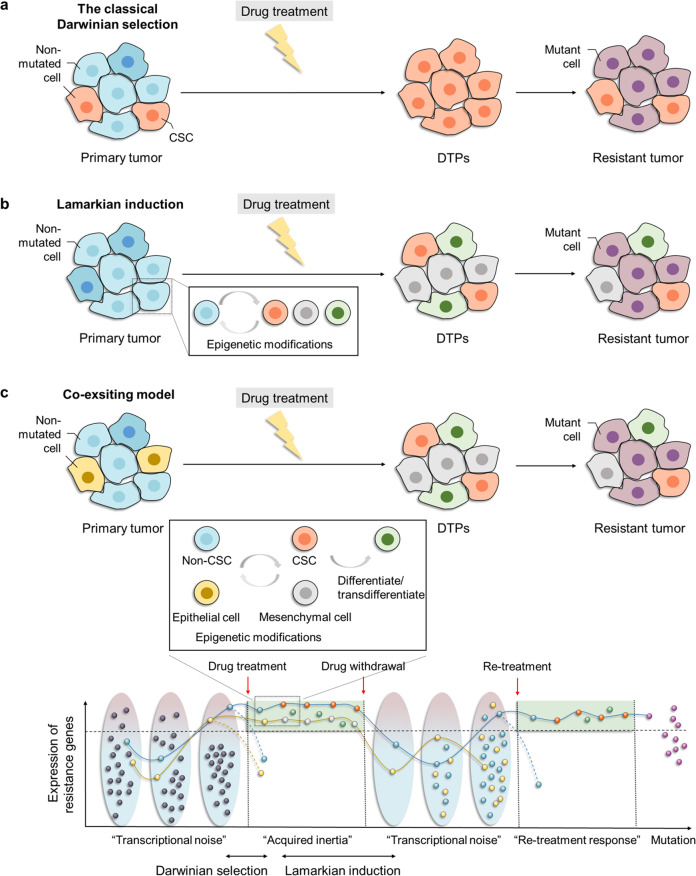
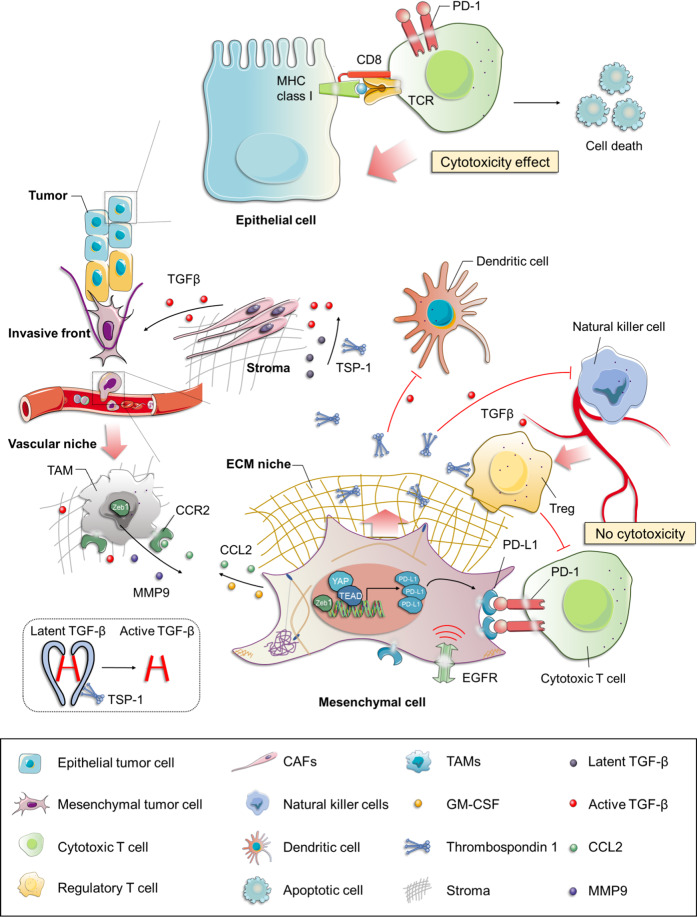
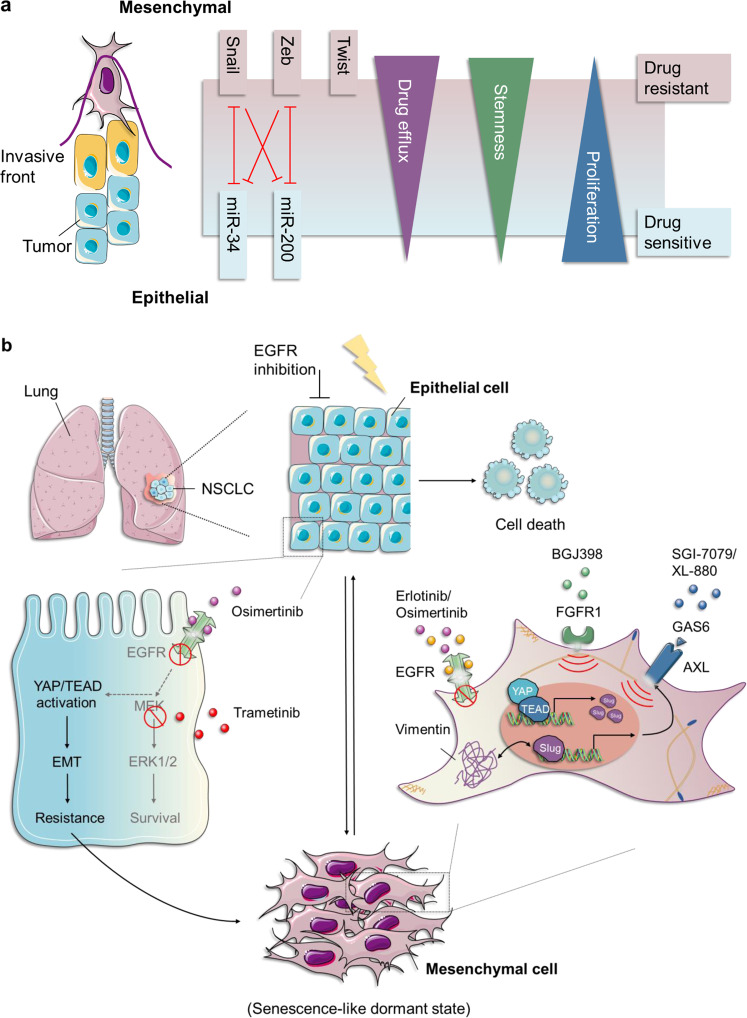
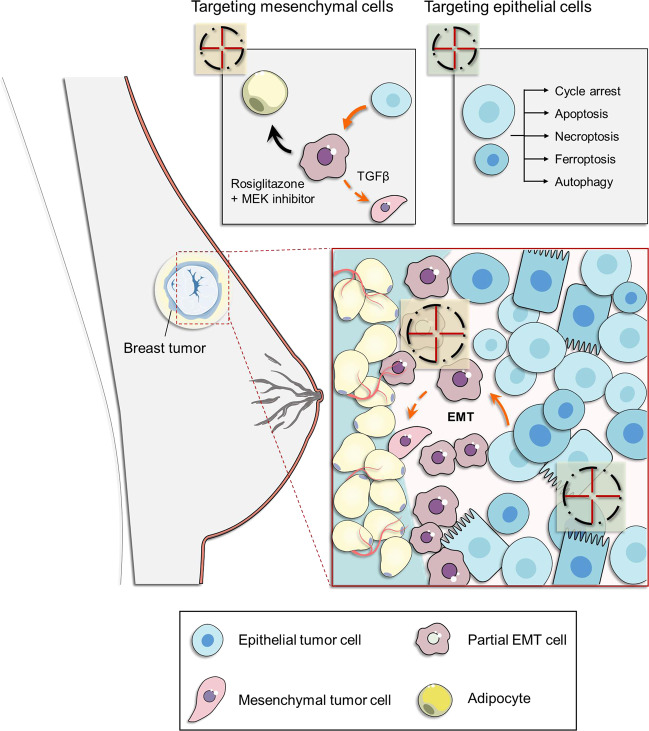
Resistance to cancer therapy is a major barrier to cancer management. Conventional views have proposed that acquisition of resistance may result from genetic mutations. However, accumulating evidence implicates a key role of non-mutational resistance mechanisms underlying drug tolerance, the latter of which is the focus that will be discussed here. Such non-mutational processes are largely driven by tumor cell plasticity, which renders tumor cells insusceptible to the drug-targeted pathway, thereby facilitating the tumor cell survival and growth. The concept of tumor cell plasticity highlights the significance of re-activation of developmental programs that are closely correlated with epithelial-mesenchymal transition, acquisition properties of cancer stem cells, and trans-differentiation potential during drug exposure. From observations in various cancers, this concept provides an opportunity for investigating the nature of anticancer drug resistance. Over the years, our understanding of the emerging role of phenotype switching in modifying therapeutic response has considerably increased. This expanded knowledge of tumor cell plasticity contributes to developing novel therapeutic strategies or combination therapy regimens using available anticancer drugs, which are likely to improve patient outcomes in clinical practice.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
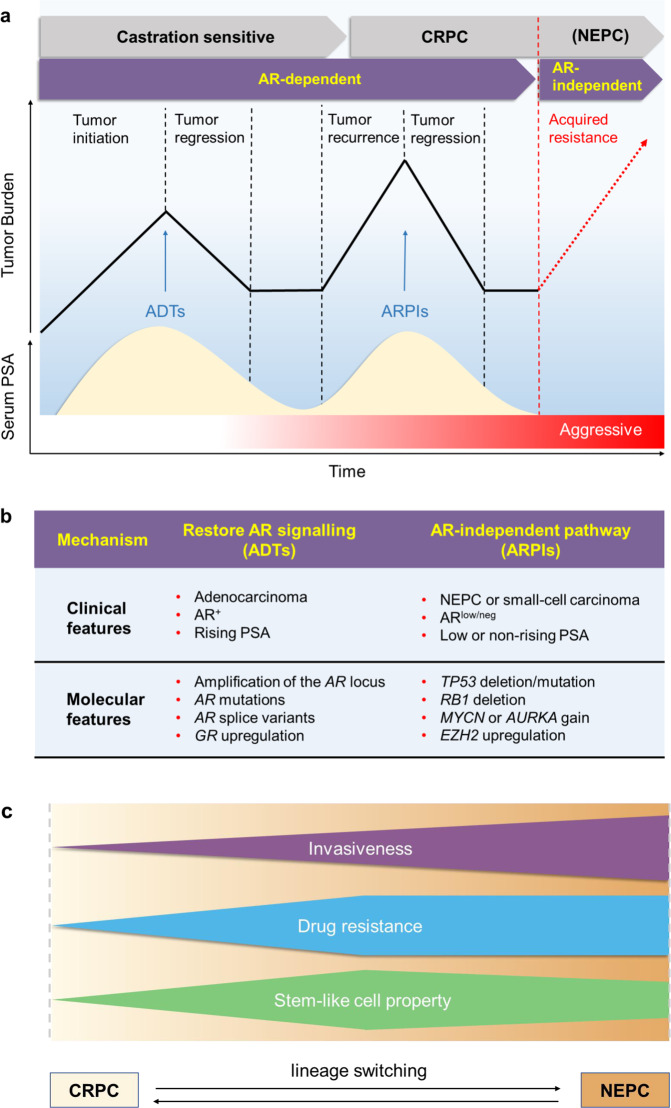
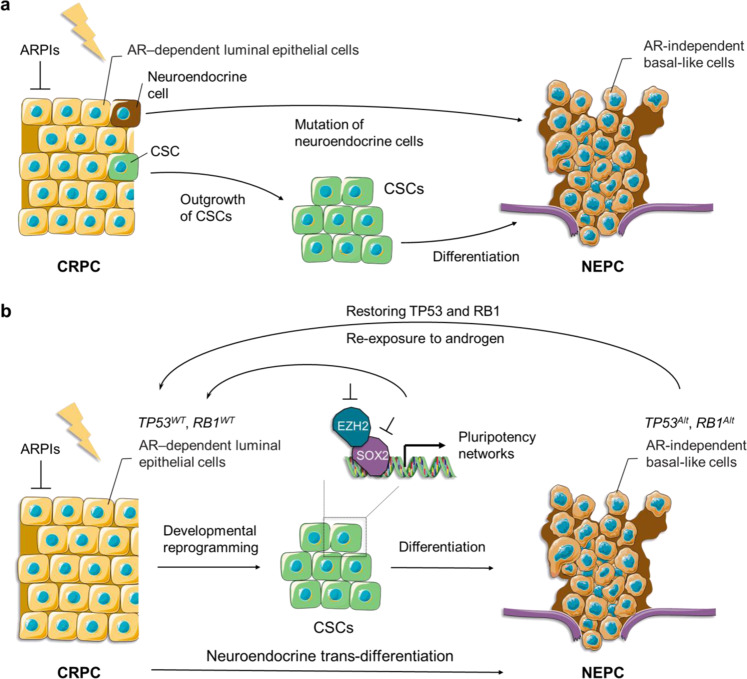
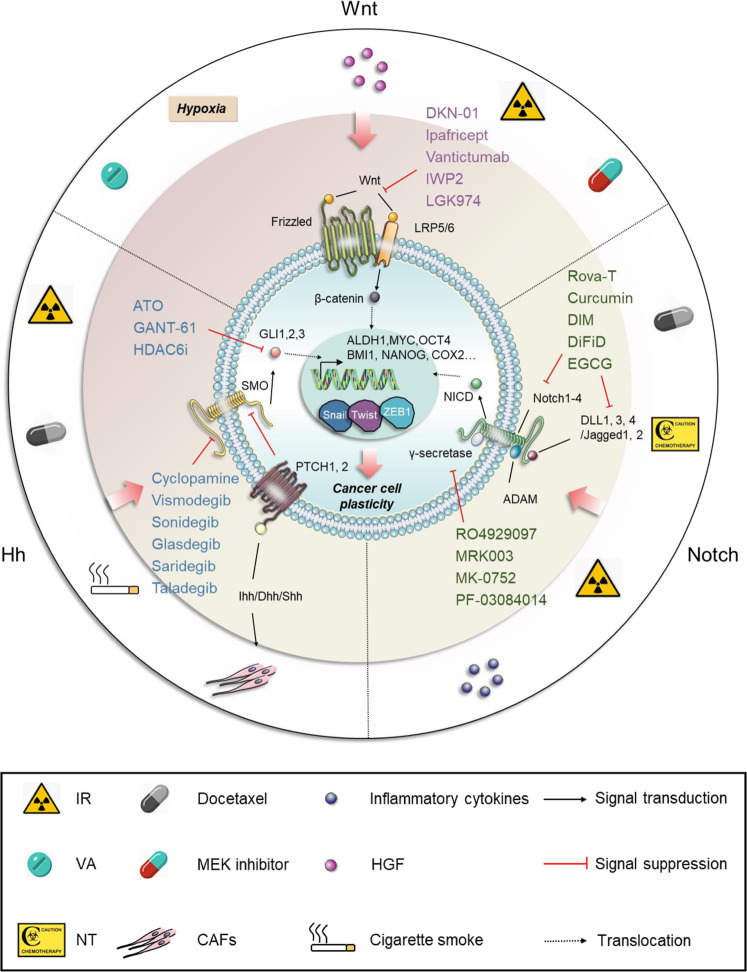
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
