

Wnt antagonist FRZB is a muscle biomarker of denervation atrophy in amyotrophic lateral sclerosis
- PMID: 33028902
- PMCID: PMC7541525
- DOI: 10.1038/s41598-020-73845-z
Wnt antagonist FRZB is a muscle biomarker of denervation atrophy in amyotrophic lateral sclerosis
Abstract
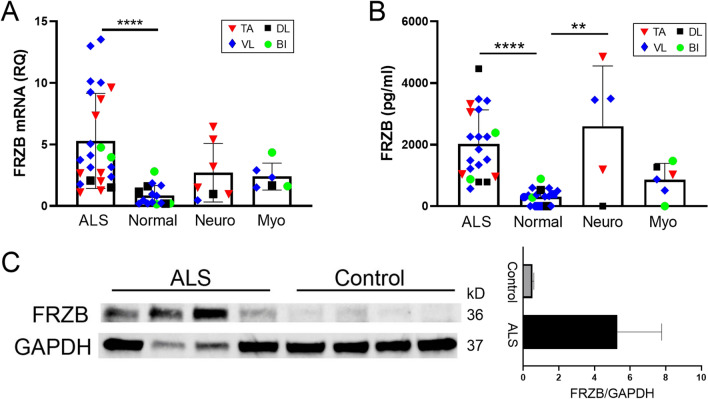
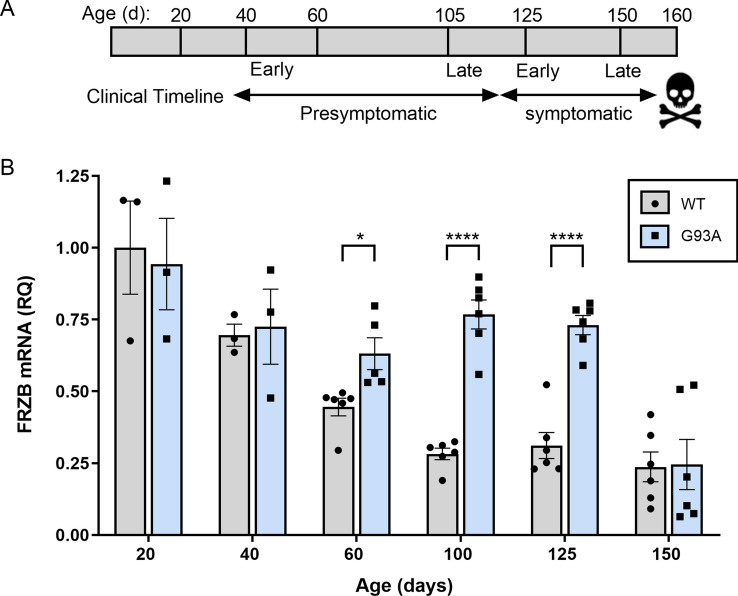
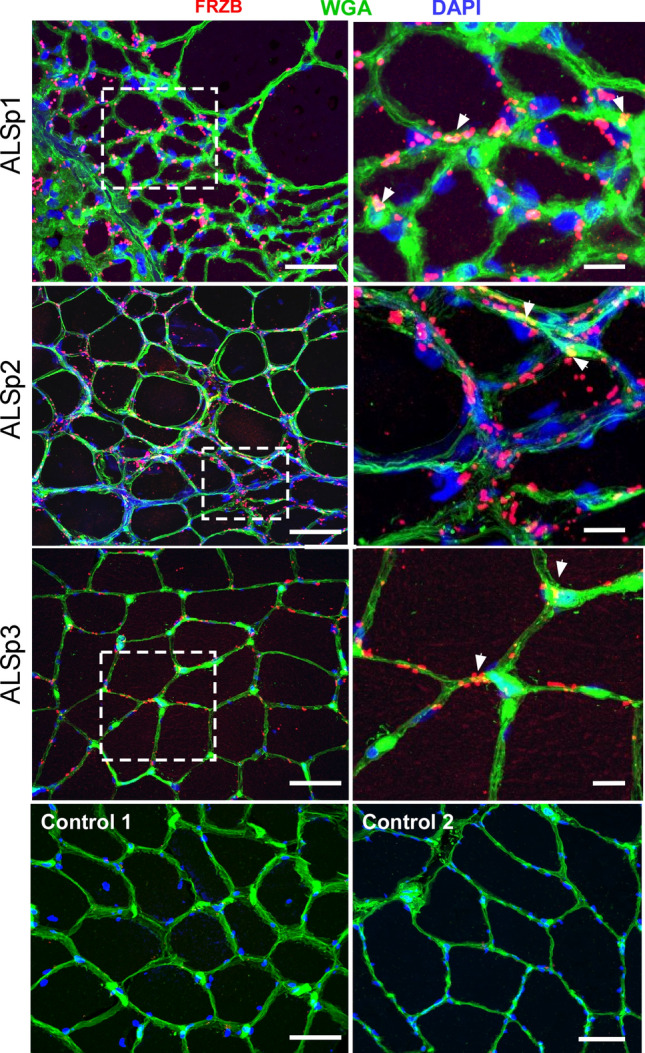
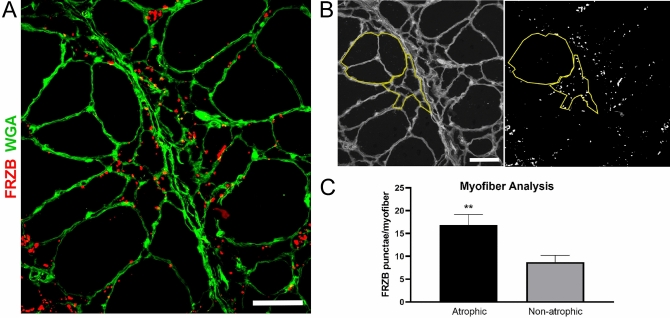
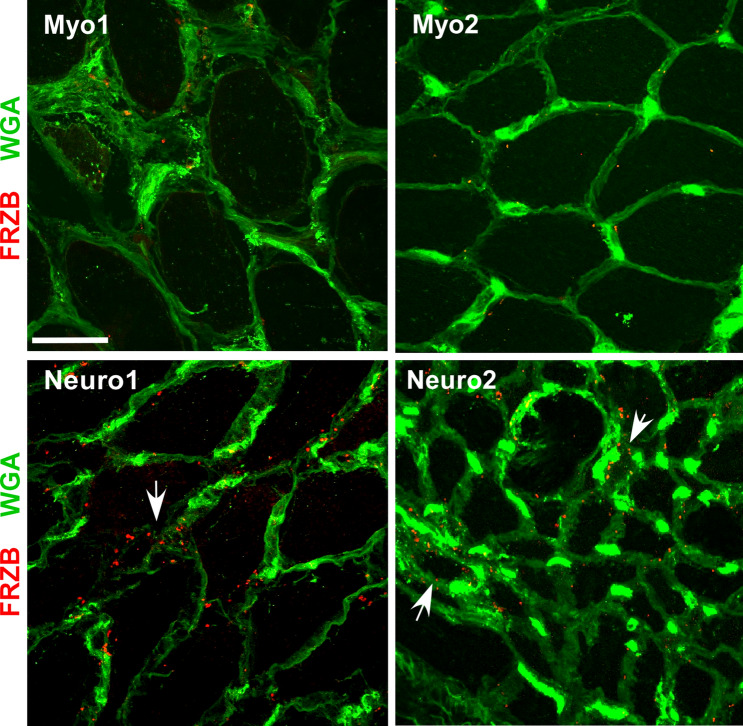
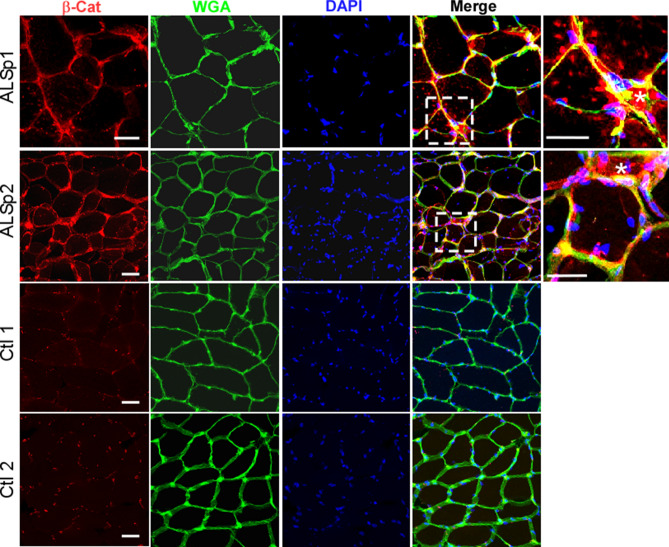
Skeletal muscle and the neuromuscular junction are the earliest sites to manifest pathological changes in amyotrophic lateral sclerosis (ALS). Based on prior studies, we have identified a molecular signature in muscle that develops early in ALS and parallels disease progression. This signature represents an intersection of signaling pathways including Smads, TGF-β, and vitamin D. Here, we show that the Wnt antagonist, Frizzled Related Protein (FRZB), was increased in ALS muscle samples and to a variable extent other denervating disease but only minimally in acquired myopathies. In the SOD1G93A mouse, FRZB was upregulated in the early stages of disease (between 40 and 60 days) until end-stage. By immunohistochemistry, FRZB was predominantly localized to endomysial connective tissue and to a lesser extent muscle membrane. There was a significant increase in immunoreactivity surrounding atrophied myofibers. Because FRZB is a Wnt antagonist, we assessed β-catenin, the canonical transducer of Wnt signaling, and found increased levels mainly at the muscle membrane. In summary, we show that FRZB is part of a molecular signature of muscle denervation that may reflect disease progression in ALS. Our findings open up avenues for future investigation as to what roles FRZB and Wnt signaling might be playing in muscle denervation/reinnervation.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
