

The genomic landscapes of individual melanocytes from human skin
- PMID: 33029006
- PMCID: PMC7581540
- DOI: 10.1038/s41586-020-2785-8
The genomic landscapes of individual melanocytes from human skin
Erratum in
-
Publisher Correction: The genomic landscapes of individual melanocytes from human skin.Nature. 2021 Feb;590(7844):E20. doi: 10.1038/s41586-020-03105-7. Nature. 2021. PMID: 33469220 No abstract available.
Abstract
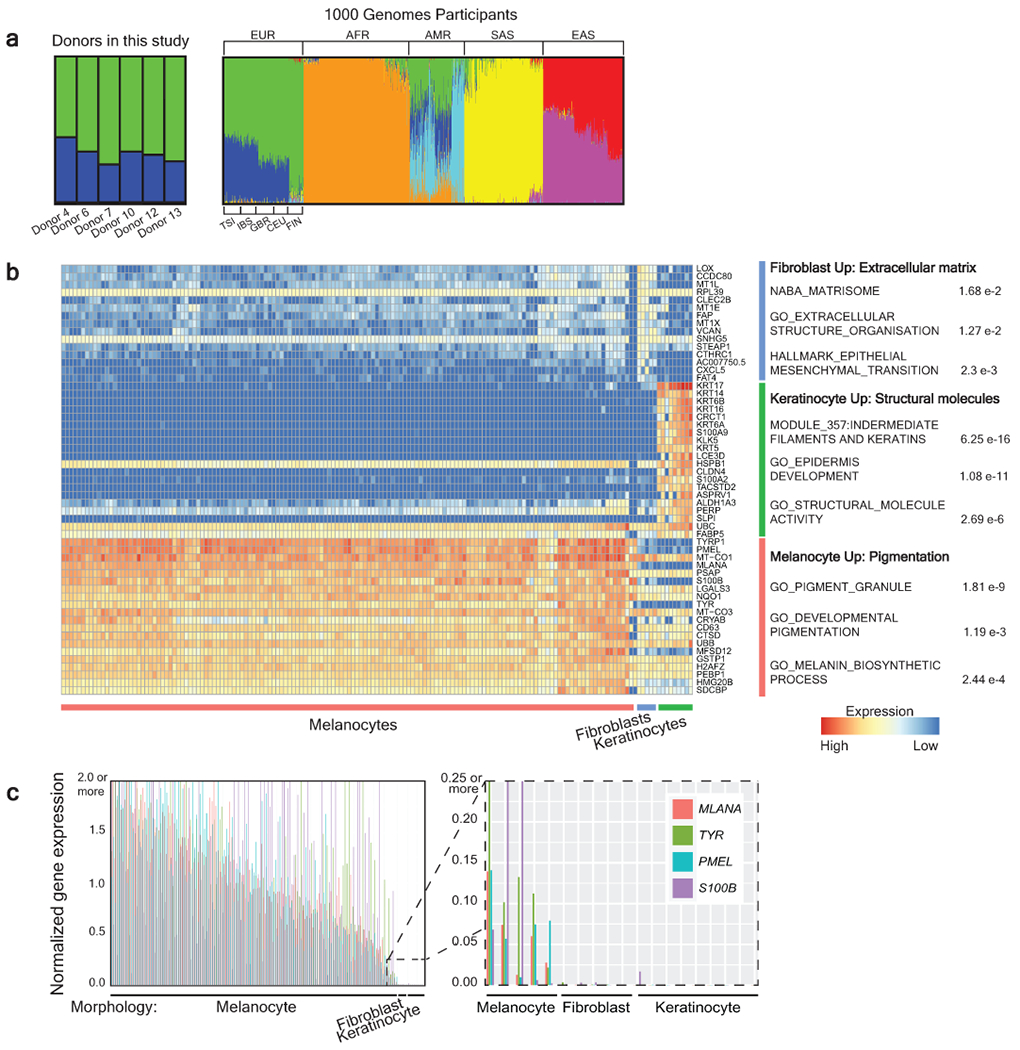
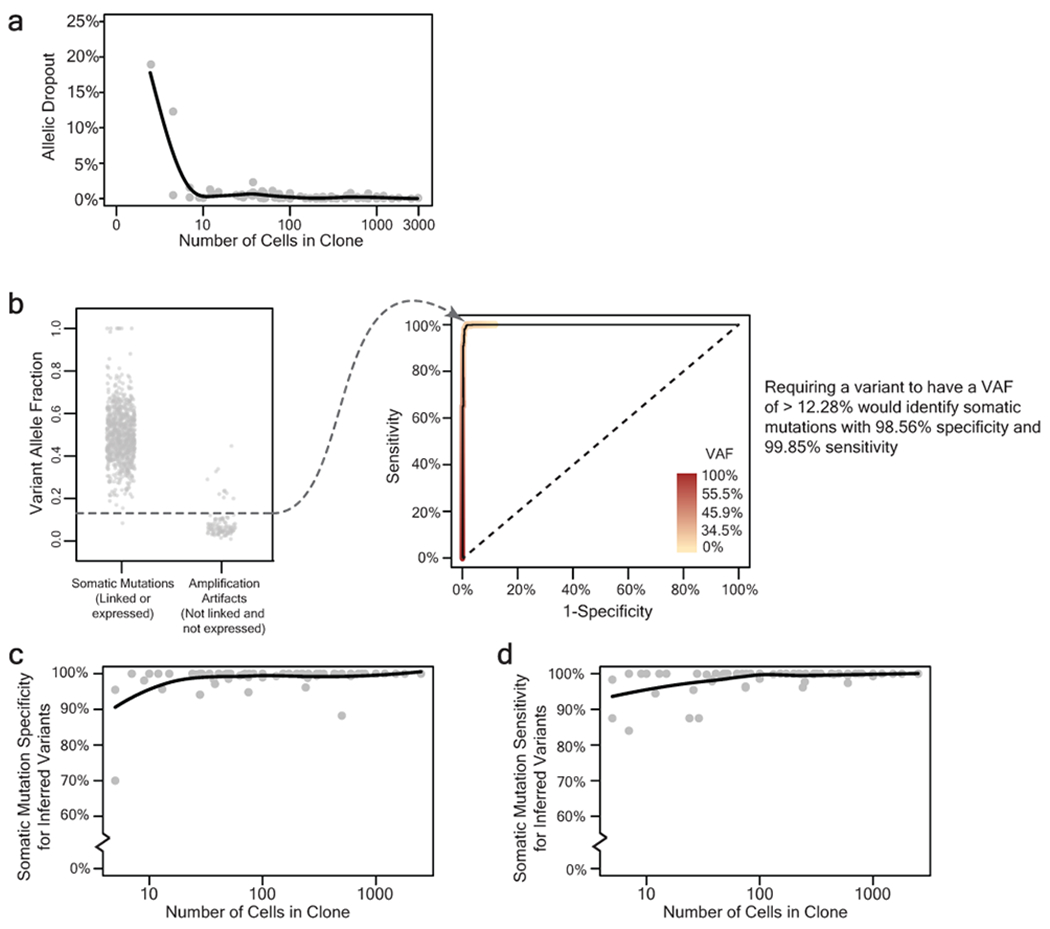
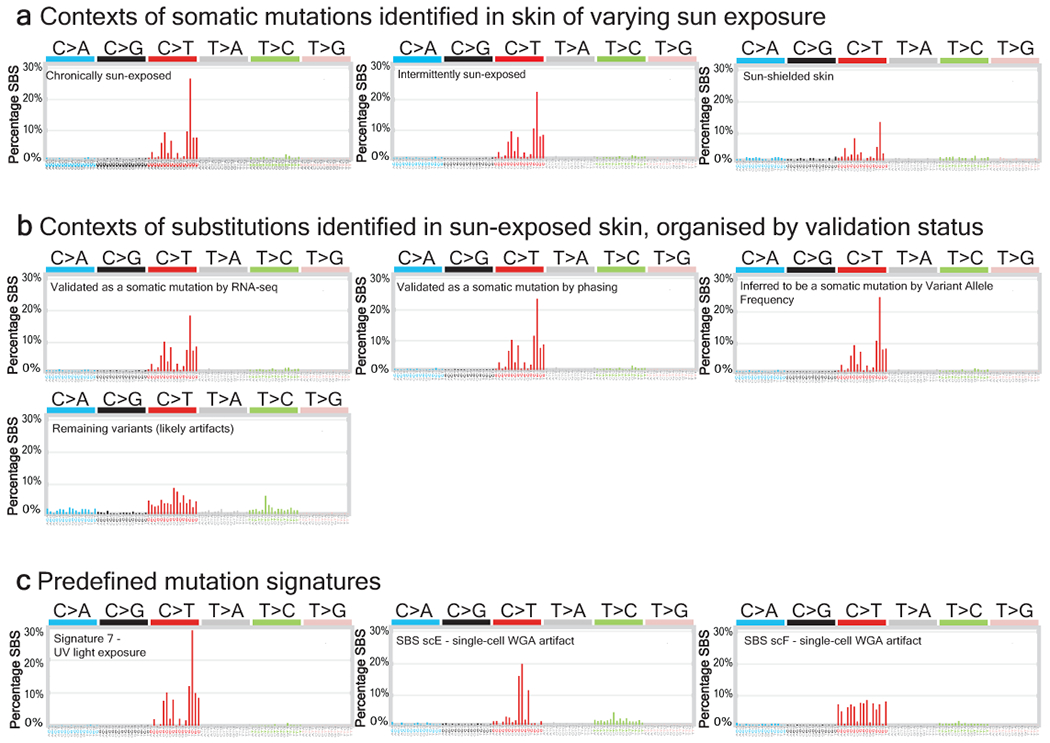
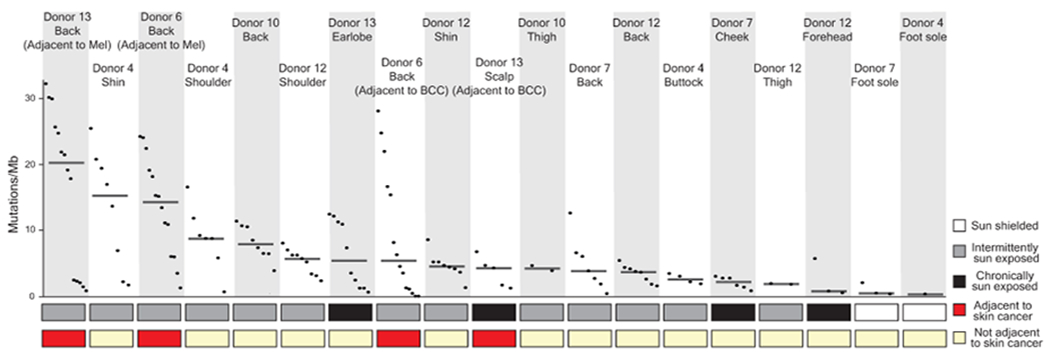
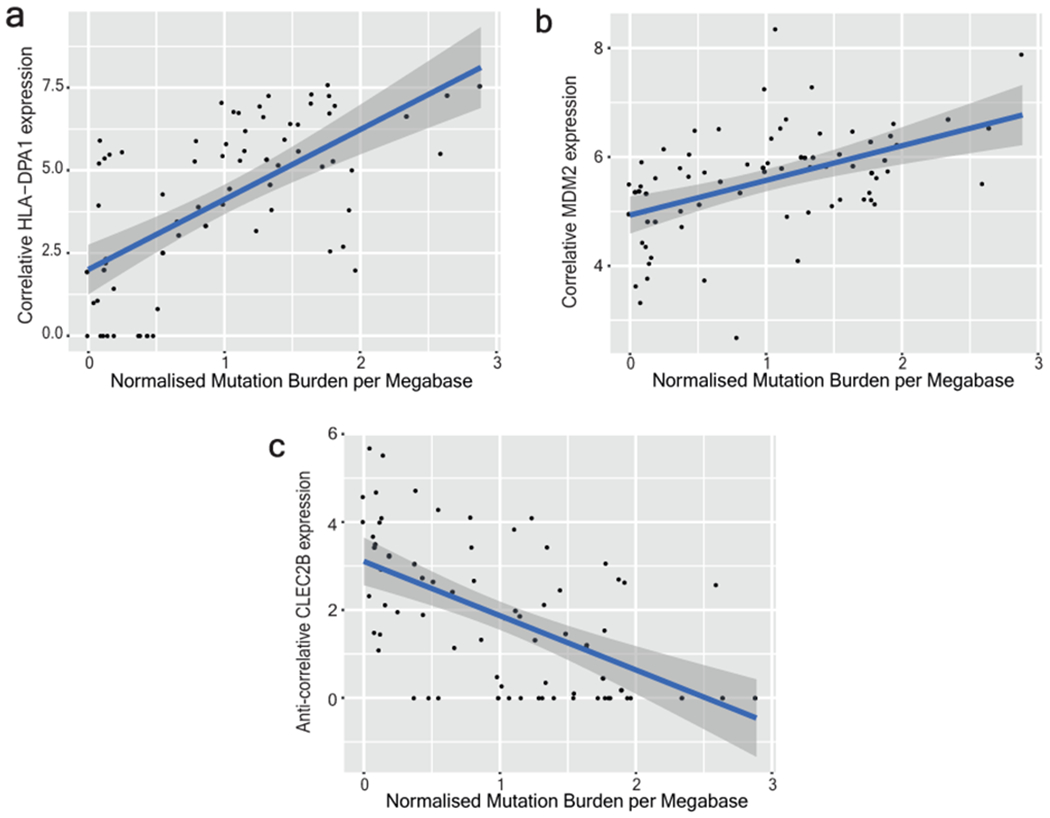
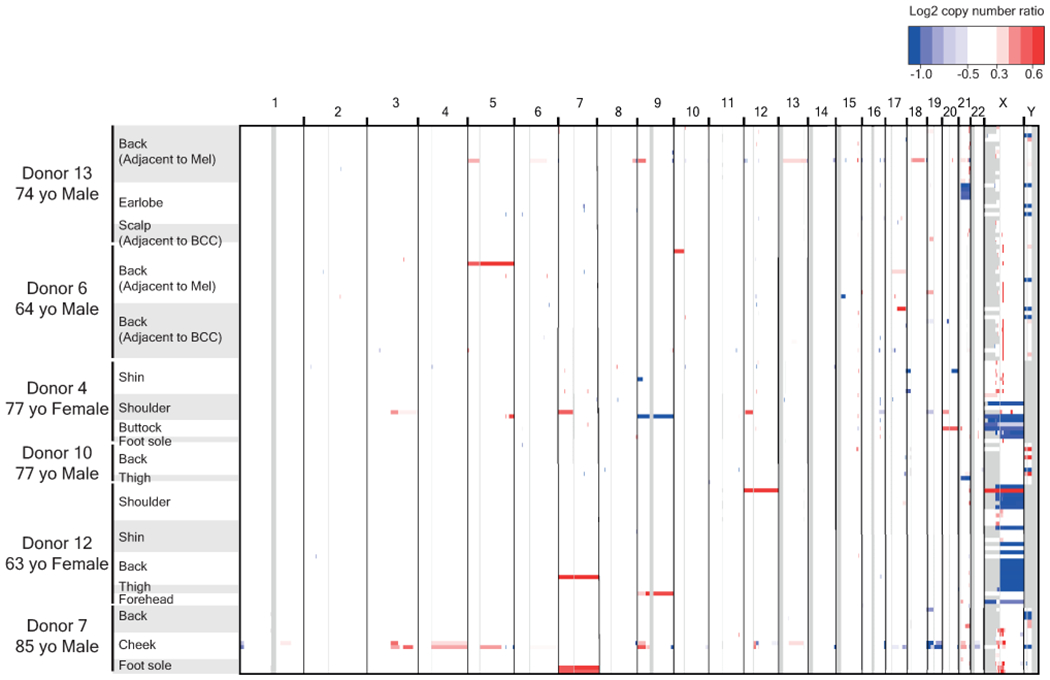
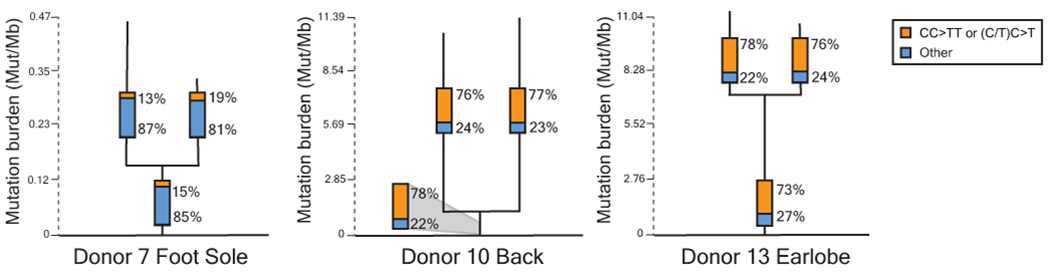
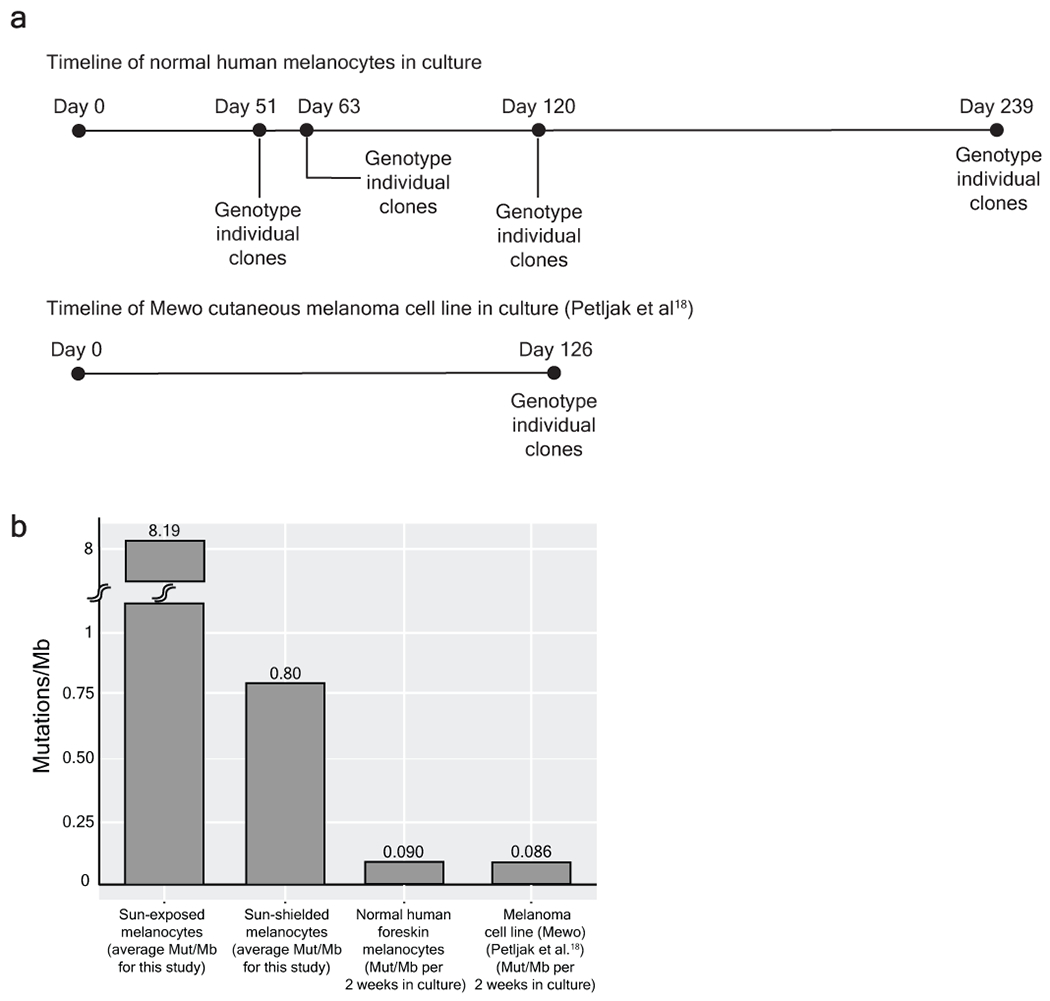
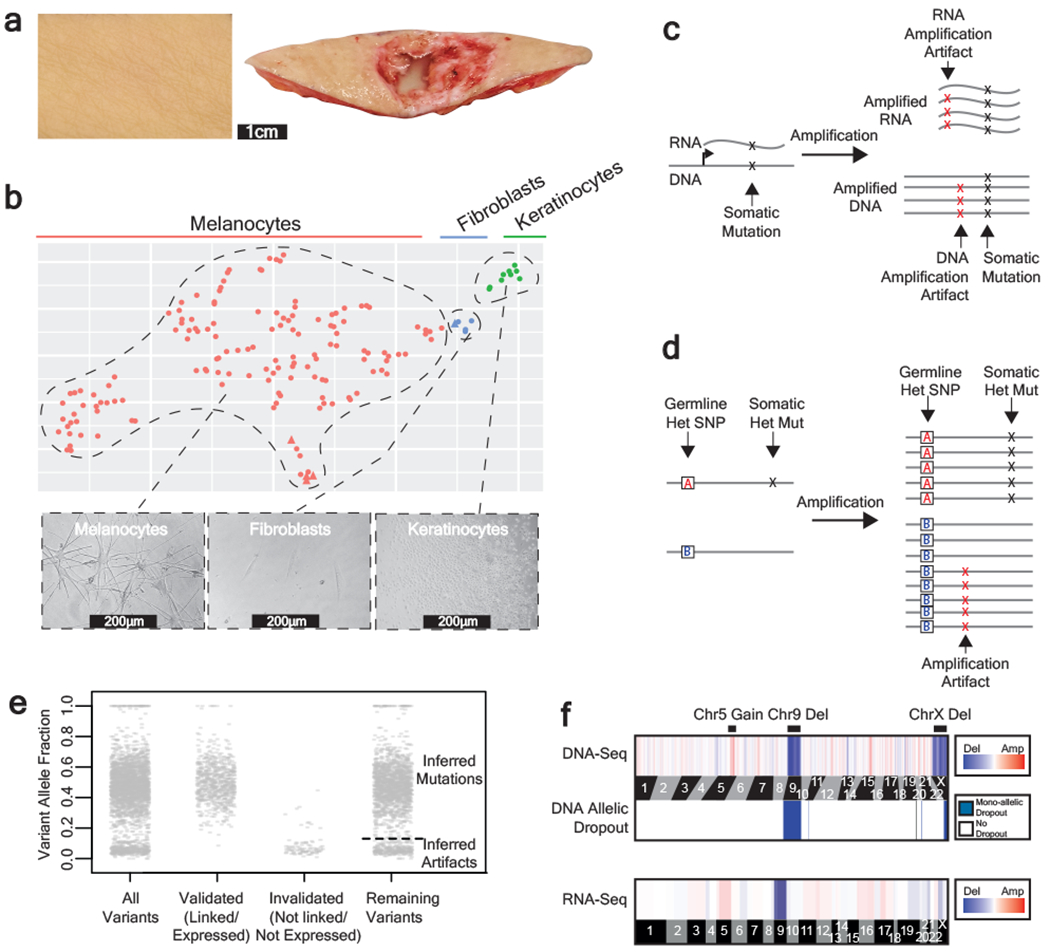
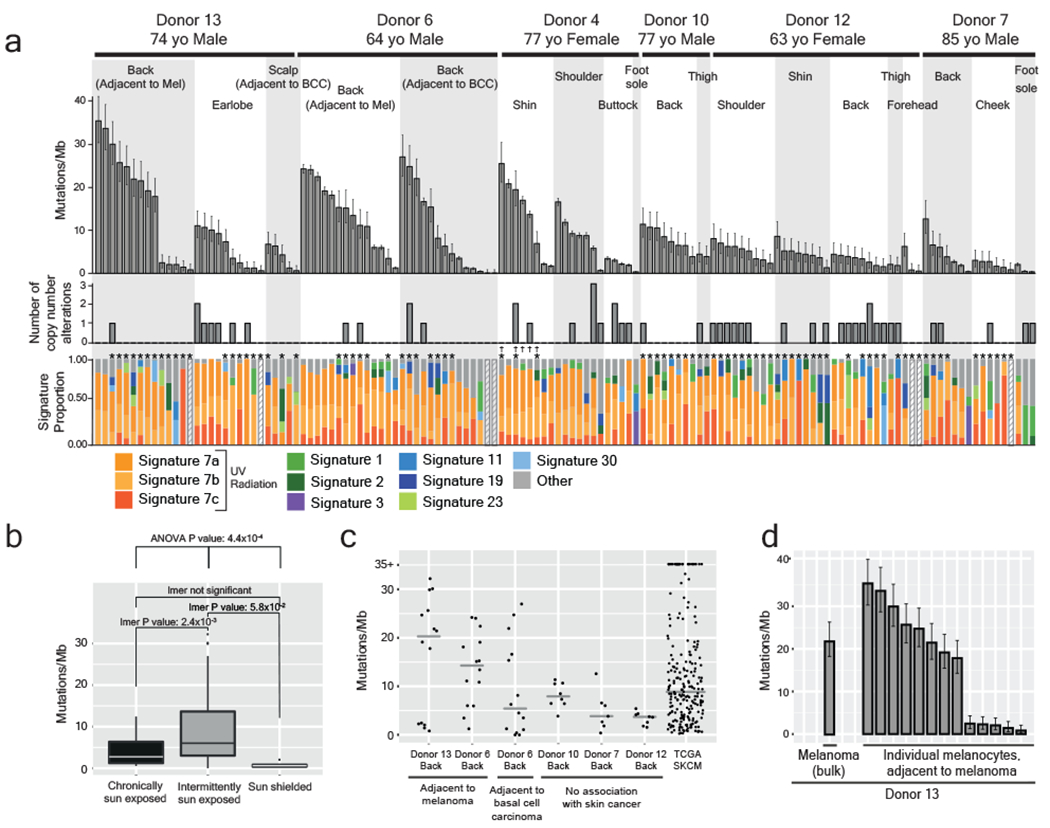
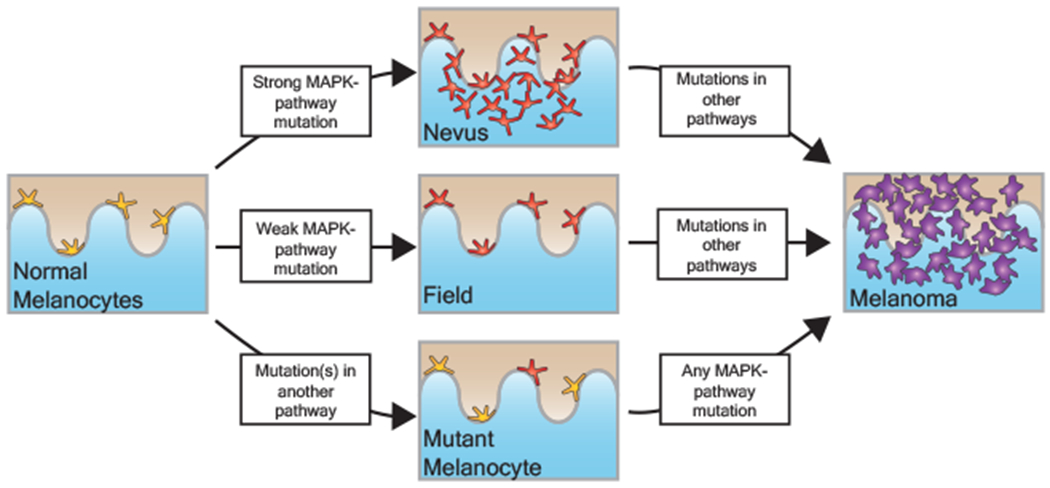
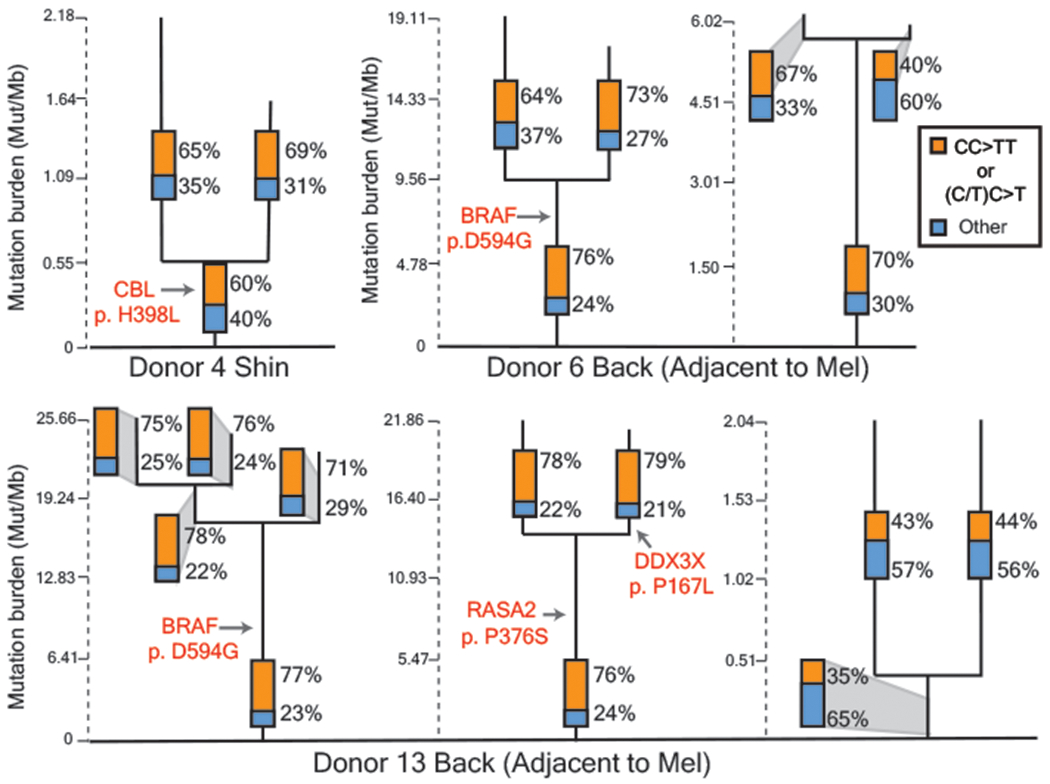
Every cell in the human body has a unique set of somatic mutations, but it remains difficult to comprehensively genotype an individual cell1. Here we describe ways to overcome this obstacle in the context of normal human skin, thus offering a glimpse into the genomic landscapes of individual melanocytes from human skin. As expected, sun-shielded melanocytes had fewer mutations than sun-exposed melanocytes. However, melanocytes from chronically sun-exposed skin (for example, the face) had a lower mutation burden than melanocytes from intermittently sun-exposed skin (for example, the back). Melanocytes located adjacent to a skin cancer had higher mutation burdens than melanocytes from donors without skin cancer, implying that the mutation burden of normal skin can be used to measure cumulative sun damage and risk of skin cancer. Moreover, melanocytes from healthy skin commonly contained pathogenic mutations, although these mutations tended to be weakly oncogenic, probably explaining why they did not give rise to discernible lesions. Phylogenetic analyses identified groups of related melanocytes, suggesting that melanocytes spread throughout skin as fields of clonally related cells that are invisible to the naked eye. Overall, our results uncover the genomic landscapes of individual melanocytes, providing key insights into the causes and origins of melanoma.
Conflict of interest statement
Competing Interests Declaration
STA is an employee at Rakuten Medical and a consultant for Castle Biosciences and Enspectra Health.
Figures
Comment in
-
Seeds of cancer in normal skin.Nature. 2020 Oct;586(7830):504-506. doi: 10.1038/d41586-020-02749-9. Nature. 2020. PMID: 33028991 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
