

Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding
- PMID: 33029559
- PMCID: PMC7454405
- DOI: 10.1093/biomethods/bpaa014
Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding
Abstract
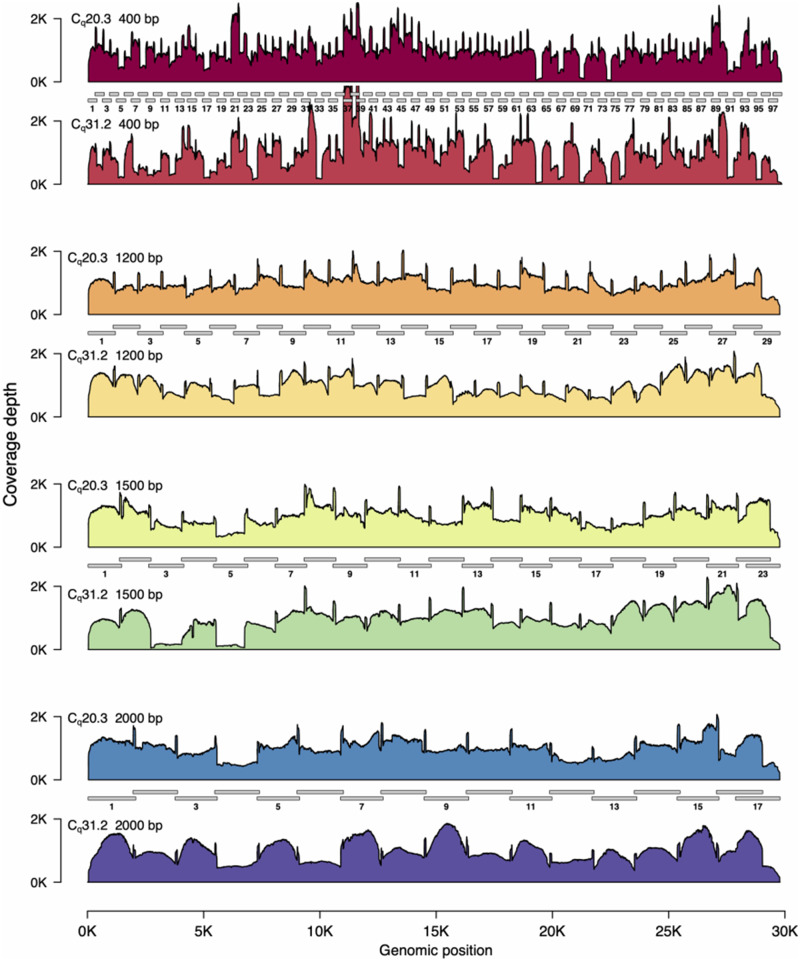
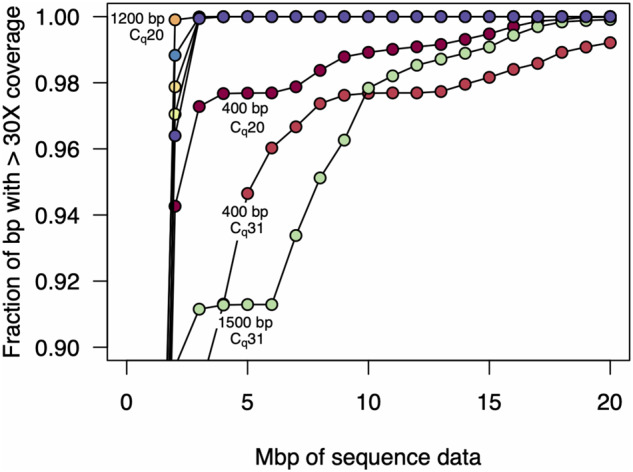
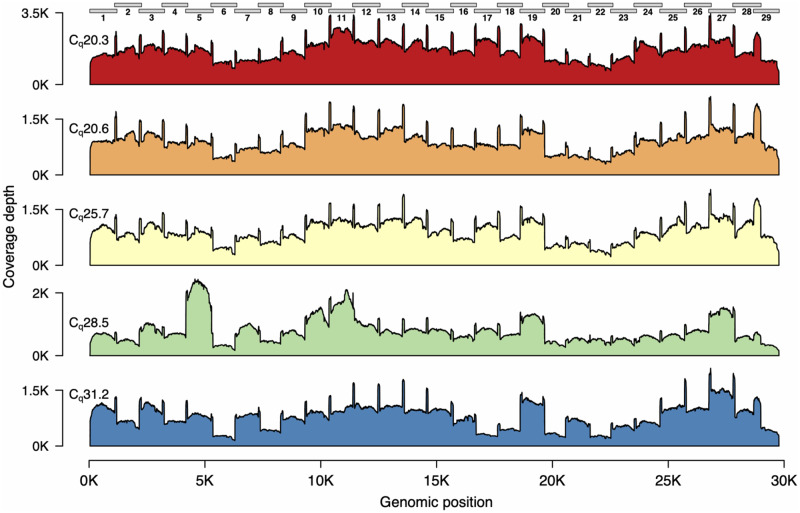
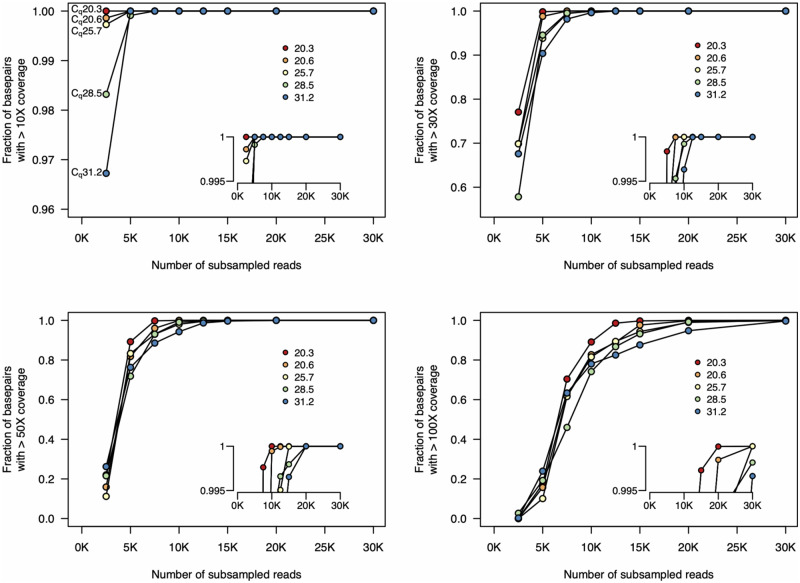
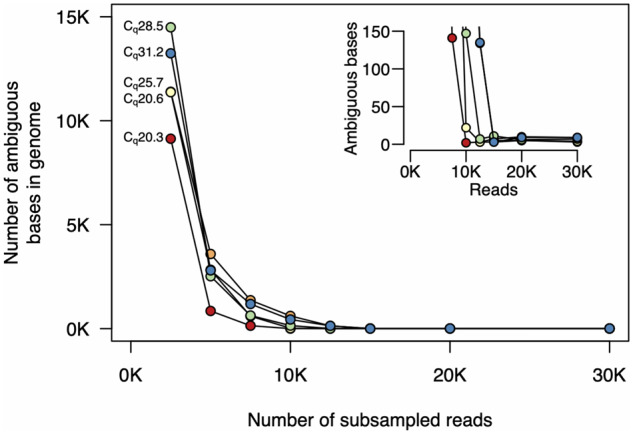
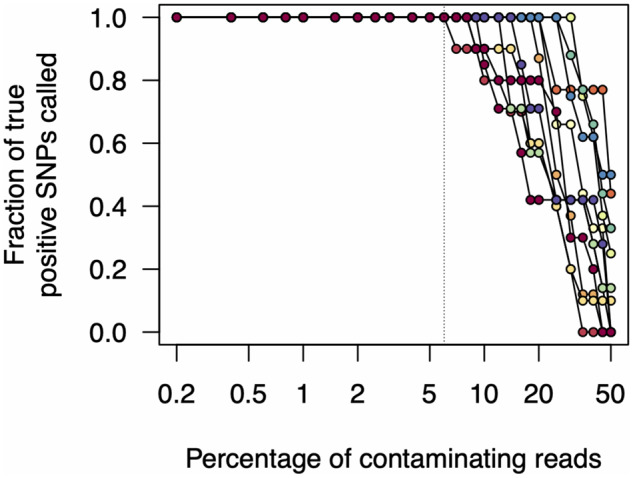
Rapid and cost-efficient whole-genome sequencing of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes coronavirus disease 2019, is critical for understanding viral transmission dynamics. Here we show that using a new multiplexed set of primers in conjunction with the Oxford Nanopore Rapid Barcode library kit allows for faster, simpler, and less expensive SARS-CoV-2 genome sequencing. This primer set results in amplicons that exhibit lower levels of variation in coverage compared to other commonly used primer sets. Using five SARS-CoV-2 patient samples with Cq values between 20 and 31, we show that high-quality genomes can be generated with as few as 10 000 reads (∼5 Mbp of sequence data). We also show that mis-classification of barcodes, which may be more likely when using the Oxford Nanopore Rapid Barcode library prep, is unlikely to cause problems in variant calling. This method reduces the time from RNA to genome sequence by more than half compared to the more standard ligation-based Oxford Nanopore library preparation method at considerably lower costs.
Keywords: Nanopore; SARS-CoV-2; amplicon; genome.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- Manning JE, Bohl JA, Lay S. et al. Rapid metagenomic characterization of a case of imported COVID-19 in Cambodia. bioRxiv 2020:2020.03.02.968818.
-
- Itokawa K, Sekizuka T, Hashino M. et al. A proposal of alternative primers for the ARTIC Network’s multiplex PCR to improve coverage of SARS-CoV-2 genome sequencing. BioRxiv2020, doi: 10.1101/2020.03.10.985150.
-
- Resende PC, Motta FC, Roy S. et al. SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. bioRxiv2020, doi: 10.1101/2020.04.30.069039.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous