

Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications
- PMID: 33029758
- PMCID: PMC7541099
- DOI: 10.1007/s10753-020-01337-3
Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications
Abstract
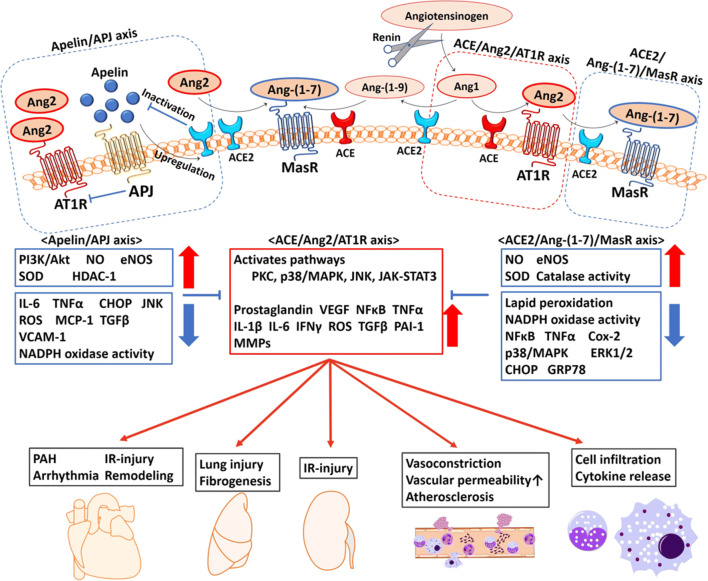
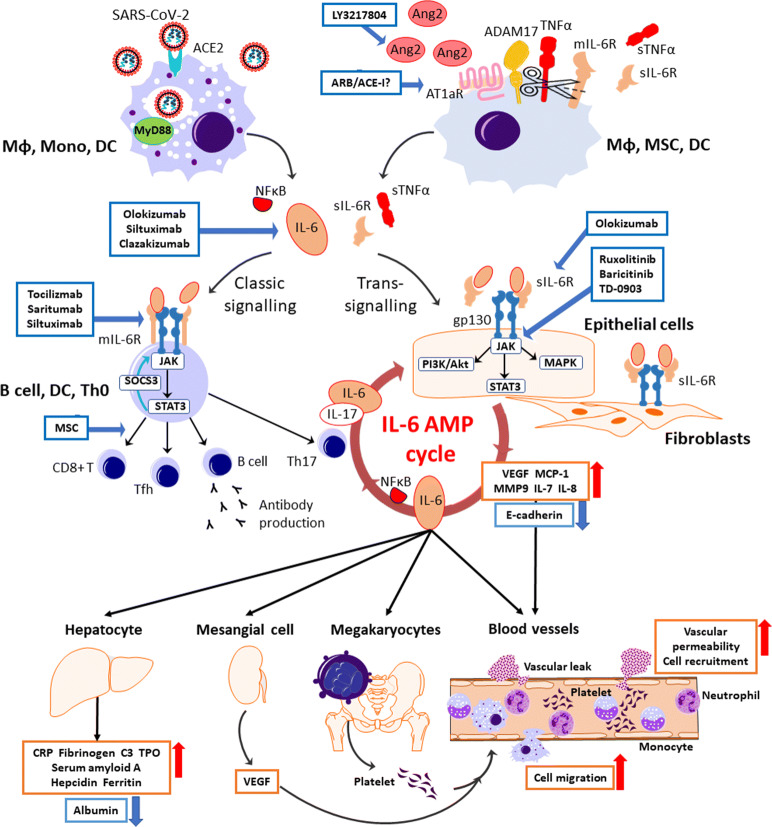
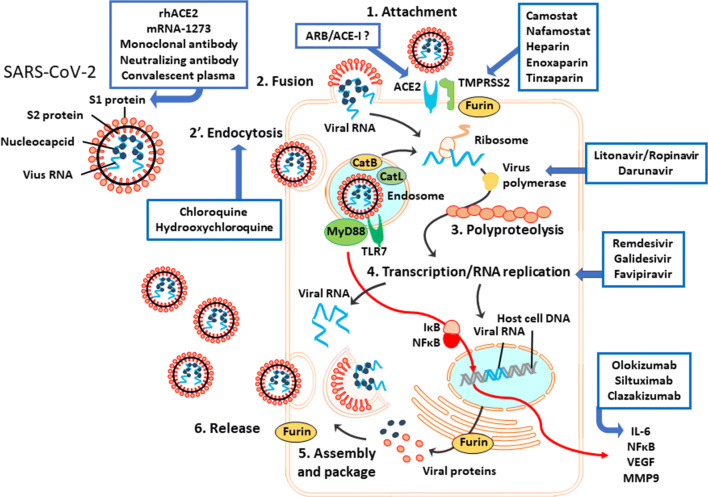
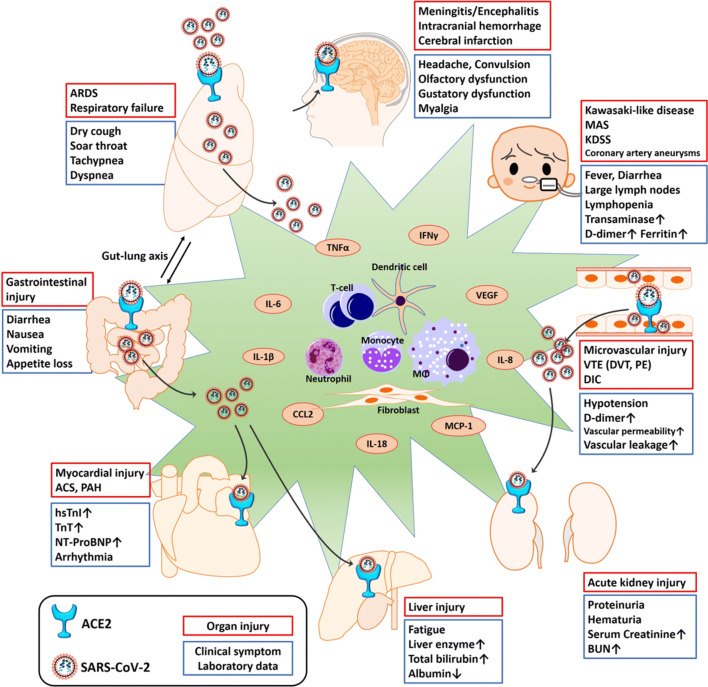
The widespread occurrence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to a pandemic of coronavirus disease 2019 (COVID-19). The S spike protein of SARS-CoV-2 binds with angiotensin-converting enzyme 2 (ACE2) as a functional "receptor" and then enters into host cells to replicate and damage host cells and organs. ACE2 plays a pivotal role in the inflammation, and its downregulation may aggravate COVID-19 via the renin-angiotensin system, including by promoting pathological changes in lung injury and involving inflammatory responses. Severe patients of COVID-19 often develop acute respiratory distress syndrome and multiple organ dysfunction/failure with high mortality that may be closely related to the hyper-proinflammatory status called the "cytokine storm." Massive cytokines including interleukin-6, nuclear factor kappa B (NFκB), and tumor necrosis factor alpha (TNFα) released from SARS-CoV-2-infected macrophages and monocytes lead inflammation-derived injurious cascades causing multi-organ injury/failure. This review summarizes the current evidence and understanding of the underlying mechanisms of SARS-CoV-2, ACE2 and inflammation co-mediated multi-organ injury or failure in COVID-19 patients.
Keywords: COVID-19; SARS-CoV-2; angiotensin-converting enzyme 2; cytokine storm; multiple organ failure; renin-angiotensin system.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Anguiano L, Riera M, Pascual J, Valdivielso JM, Barrios C, Betriu A, Mojal S, Fernandez E, Soler MJ, Nefrona study Circulating angiotensin-converting enzyme 2 activity in patients with chronic kidney disease without previous history of cardiovascular disease. Nephrology, Dialysis, Transplantation. 2015;30(7):1176–1185. doi: 10.1093/ndt/gfv025. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
