

A Switch between Two Intrinsically Disordered Conformational Ensembles Modulates the Active Site of a Basic-Helix-Loop-Helix Transcription Factor
- PMID: 33030907
- PMCID: PMC7649839
- DOI: 10.1021/acs.jpclett.0c02242
A Switch between Two Intrinsically Disordered Conformational Ensembles Modulates the Active Site of a Basic-Helix-Loop-Helix Transcription Factor
Abstract
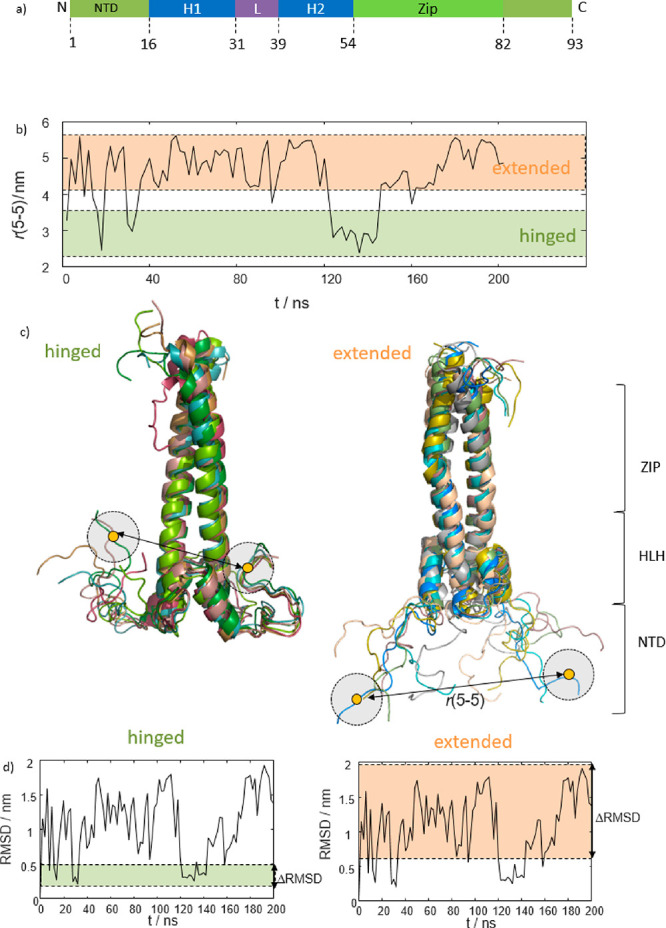
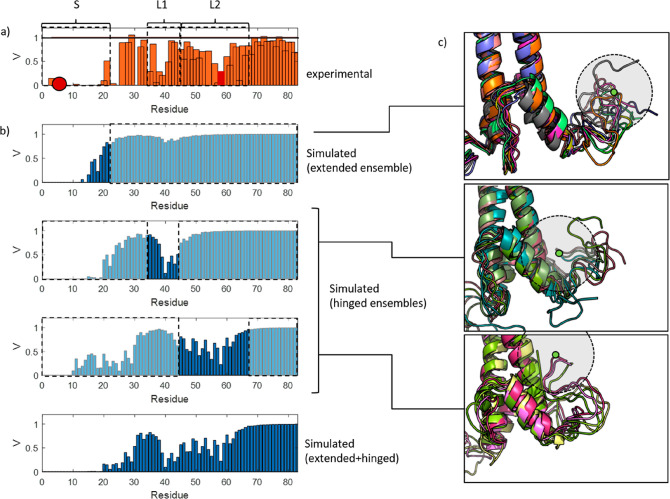
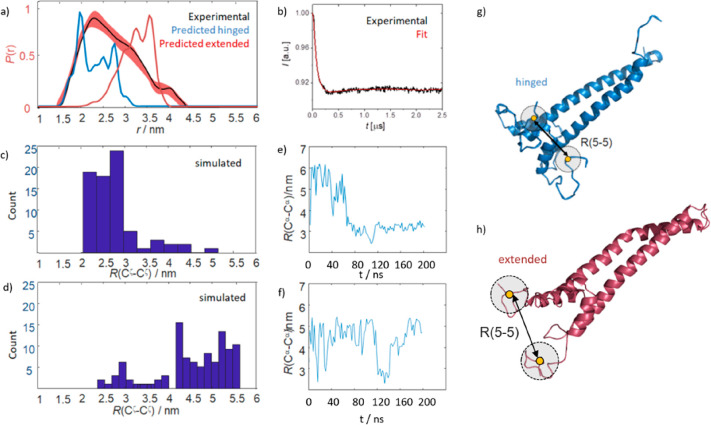
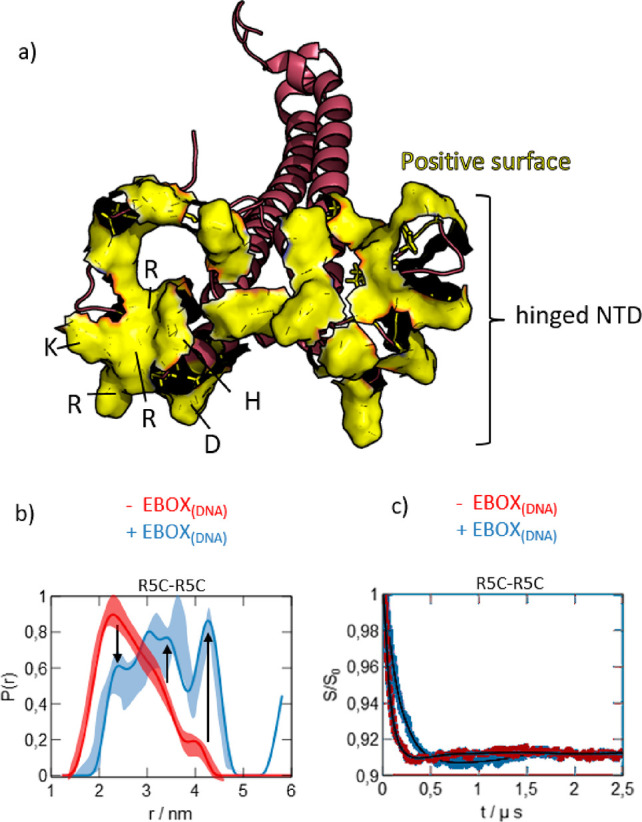
We report a conformational switch between two distinct intrinsically disordered subensembles within the active site of a transcription factor. This switch highlights an evolutionary benefit conferred by the high plasticity of intrinsically disordered domains, namely, their potential to dynamically sample a heterogeneous conformational space housing multiple states with tailored properties. We focus on proto-oncogenic basic-helix-loop-helix (bHLH)-type transcription factors, as these play key roles in cell regulation and function. Despite intense research efforts, the understanding of structure-function relations of these transcription factors remains incomplete as they feature intrinsically disordered DNA-interaction domains that are difficult to characterize, theoretically as well as experimentally. Here we characterize the structural dynamics of the intrinsically disordered region DNA-binding site of the vital MYC-associated transcription factor X (MAX). Integrating nuclear magnetic resonance (NMR) measurements, molecular dynamics (MD) simulations, and electron paramagnetic resonance (EPR) measurements, we show that, in the absence of DNA, the binding site of the free MAX2 homodimer samples two intrinsically disordered conformational subensembles. These feature distinct structural properties: one subensemble consists of a set of highly flexible and spatially extended conformers, while the second features a set of "hinged" conformations. In this latter ensemble, the disordered N-terminal tails of MAX2 fold back along the dimer, forming transient long-range contacts with the HLH-region and thereby exposing the DNA binding site to the solvent. The features of these divergent substates suggest two mechanisms by which protein conformational dynamics in MAX2 might modulate DNA-complex formation: by enhanced initial recruitment of free DNA ligands, as a result of the wider conformational space sampled by the extended ensemble, and by direct exposure of the binding site and the corresponding strong electrostatic attractions presented while in the hinged conformations.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
The NMR solution structure of a mutant of the Max b/HLH/LZ free of DNA: insights into the specific and reversible DNA binding mechanism of dimeric transcription factors.J Mol Biol. 2004 Sep 17;342(3):813-32. doi: 10.1016/j.jmb.2004.07.058. J Mol Biol. 2004. PMID: 15342239
-
The crystal structure of an intact human Max-DNA complex: new insights into mechanisms of transcriptional control.Structure. 1997 Apr 15;5(4):509-20. doi: 10.1016/s0969-2126(97)00207-4. Structure. 1997. PMID: 9115440
-
Conformational Ensemble and Biological Role of the TCTP Intrinsically Disordered Region: Influence of Calcium and Phosphorylation.J Mol Biol. 2018 May 25;430(11):1621-1639. doi: 10.1016/j.jmb.2018.04.024. Epub 2018 Apr 30. J Mol Biol. 2018. PMID: 29719201
-
Molecular Dynamics Simulations Combined with Nuclear Magnetic Resonance and/or Small-Angle X-ray Scattering Data for Characterizing Intrinsically Disordered Protein Conformational Ensembles.J Chem Inf Model. 2019 May 28;59(5):1743-1758. doi: 10.1021/acs.jcim.8b00928. Epub 2019 Mar 18. J Chem Inf Model. 2019. PMID: 30840442 Review.
-
Enzyme dynamics from NMR spectroscopy.Acc Chem Res. 2015 Feb 17;48(2):457-65. doi: 10.1021/ar500340a. Epub 2015 Jan 9. Acc Chem Res. 2015. PMID: 25574774 Free PMC article. Review.
Cited by
-
How to assess the structural dynamics of transcription factors by integrating sparse NMR and EPR constraints with molecular dynamics simulations.Comput Struct Biotechnol J. 2021 Apr 21;19:2097-2105. doi: 10.1016/j.csbj.2021.04.020. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 33995905 Free PMC article. Review.
-
EPR Spectroscopy Provides New Insights into Complex Biological Reaction Mechanisms.J Phys Chem B. 2022 Oct 6;126(39):7486-7494. doi: 10.1021/acs.jpcb.2c05235. Epub 2022 Sep 22. J Phys Chem B. 2022. PMID: 36137278 Free PMC article. Review.
-
Toward protein NMR at physiological concentrations by hyperpolarized water-Finding and mapping uncharted conformational spaces.Sci Adv. 2022 Aug 5;8(31):eabq5179. doi: 10.1126/sciadv.abq5179. Epub 2022 Aug 5. Sci Adv. 2022. PMID: 35930648 Free PMC article.
-
Evidence for an Ordering Transition near 120 K in an Intrinsically Disordered Protein, Casein.Molecules. 2021 Oct 1;26(19):5971. doi: 10.3390/molecules26195971. Molecules. 2021. PMID: 34641515 Free PMC article.
References
-
- Contreras-Martos S.; Piai A.; Kosol S.; Varadi M.; Bekesi A.; Lebrun P.; Volkov A. N.; Gevaert K.; Pierattelli R.; Felli I. C.; Tompa P. Linking functions: an additional role for an intrinsically disordered linker domain in the transcriptional coactivator CBP. Sci. Rep. 2017, 7 (1), 4676.10.1038/s41598-017-04611-x. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
