

Analysis of Trans-Ancestral SLE Risk Loci Identifies Unique Biologic Networks and Drug Targets in African and European Ancestries
- PMID: 33031749
- PMCID: PMC7675009
- DOI: 10.1016/j.ajhg.2020.09.007
Analysis of Trans-Ancestral SLE Risk Loci Identifies Unique Biologic Networks and Drug Targets in African and European Ancestries
Abstract
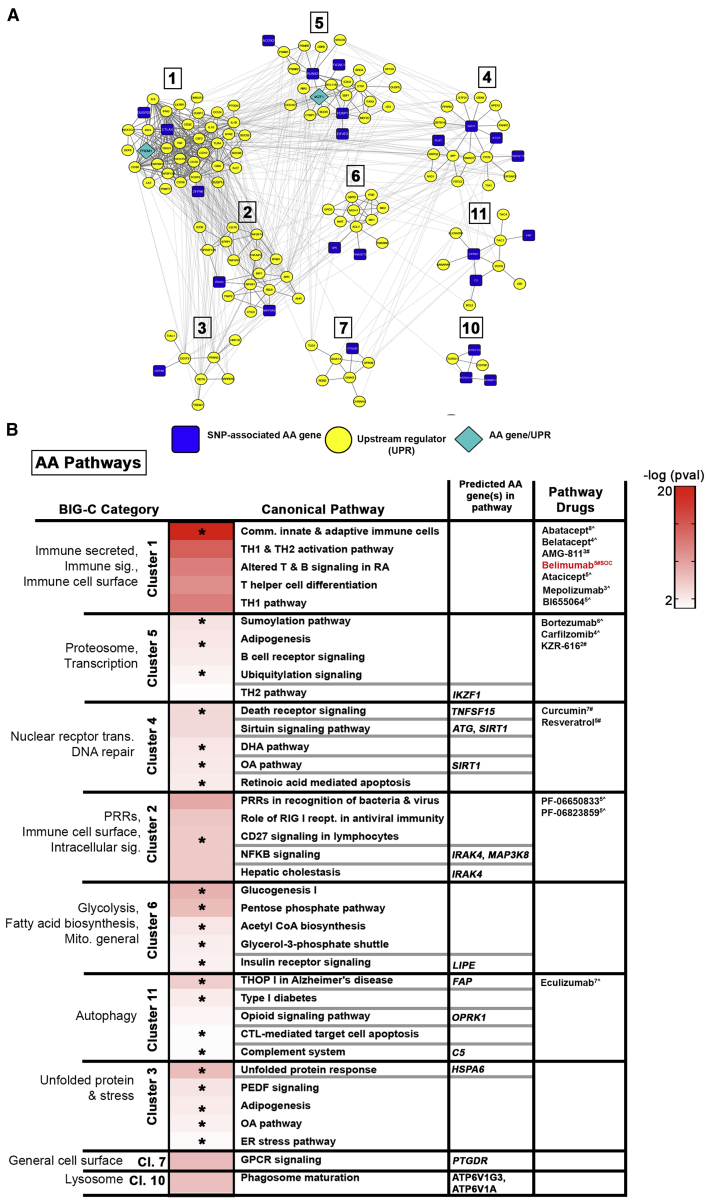
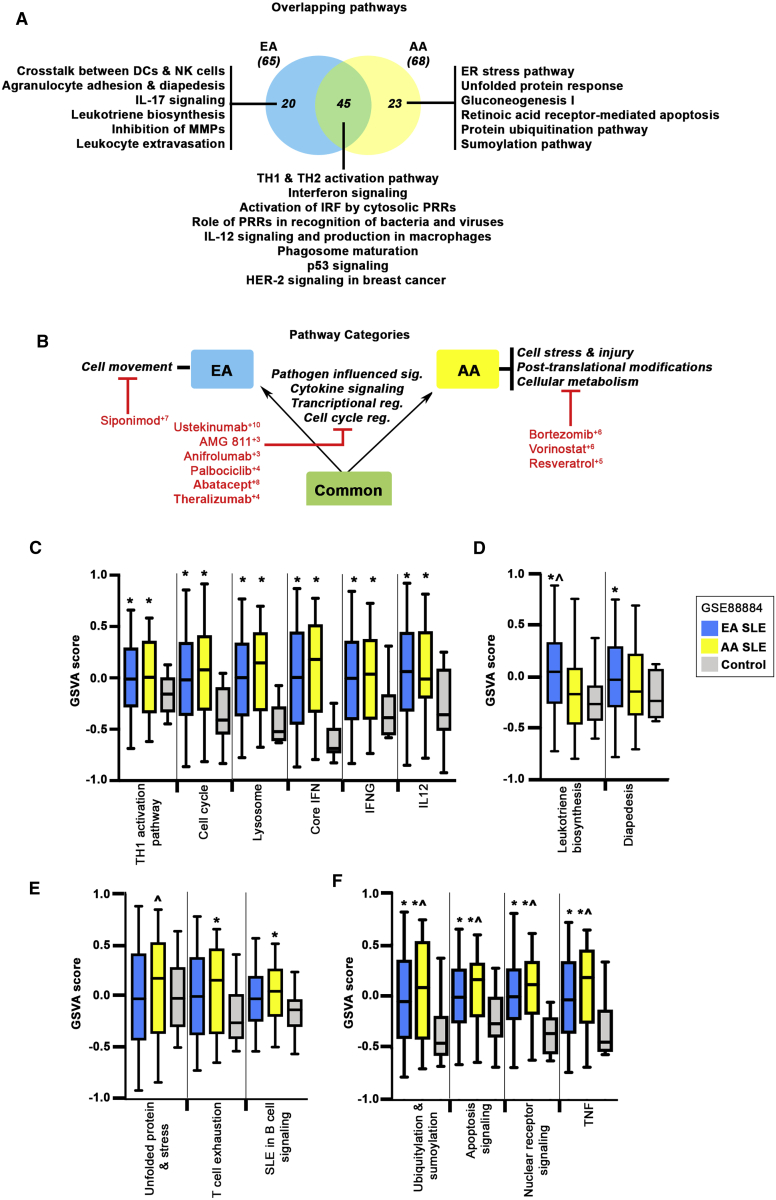
Systemic lupus erythematosus (SLE) is a multi-organ autoimmune disorder with a prominent genetic component. Individuals of African ancestry (AA) experience the disease more severely and with an increased co-morbidity burden compared to European ancestry (EA) populations. We hypothesize that the disparities in disease prevalence, activity, and response to standard medications between AA and EA populations is partially conferred by genomic influences on biological pathways. To address this, we applied a comprehensive approach to identify all genes predicted from SNP-associated risk loci detected with the Immunochip. By combining genes predicted via eQTL analysis, as well as those predicted from base-pair changes in intergenic enhancer sites, coding-region variants, and SNP-gene proximity, we were able to identify 1,731 potential ancestry-specific and trans-ancestry genetic drivers of SLE. Gene associations were linked to upstream and downstream regulators using connectivity mapping, and predicted biological pathways were mined for candidate drug targets. Examination of trans-ancestral pathways reflect the well-defined role for interferons in SLE and revealed pathways associated with tissue repair and remodeling. EA-dominant genetic drivers were more often associated with innate immune and myeloid cell function pathways, whereas AA-dominant pathways mirror clinical findings in AA subjects, suggesting disease progression is driven by aberrant B cell activity accompanied by ER stress and metabolic dysfunction. Finally, potential ancestry-specific and non-specific drug candidates were identified. The integration of all SLE SNP-predicted genes into functional pathways revealed critical molecular pathways representative of each population, underscoring the influence of ancestry on disease mechanism and also providing key insight for therapeutic selection.
Keywords: GWAS; Immunochip; SLE; ancesty; drug repurposing; genetics; lupus; pathway analysis.
Copyright © 2020 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
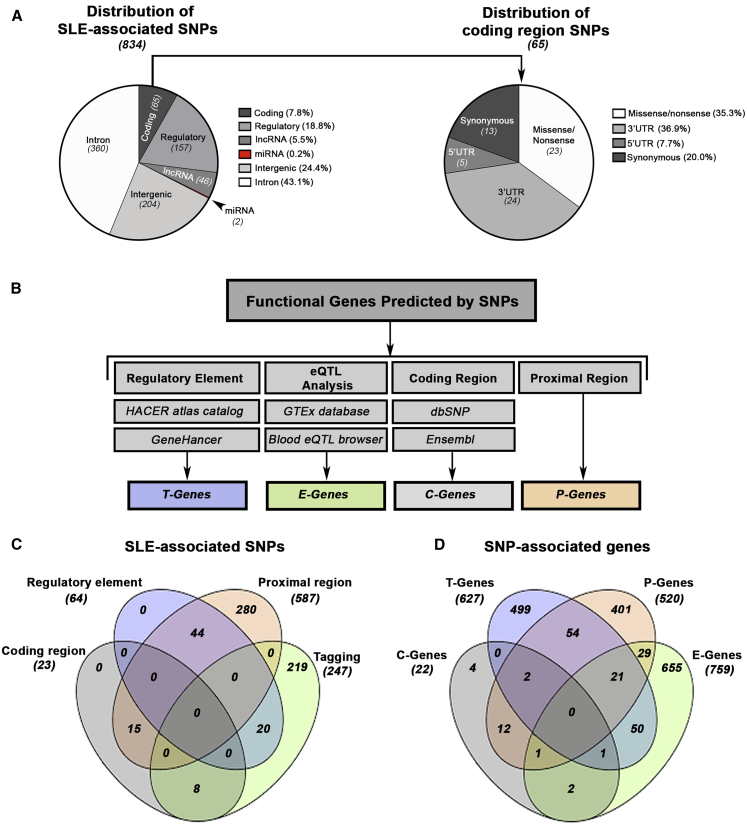
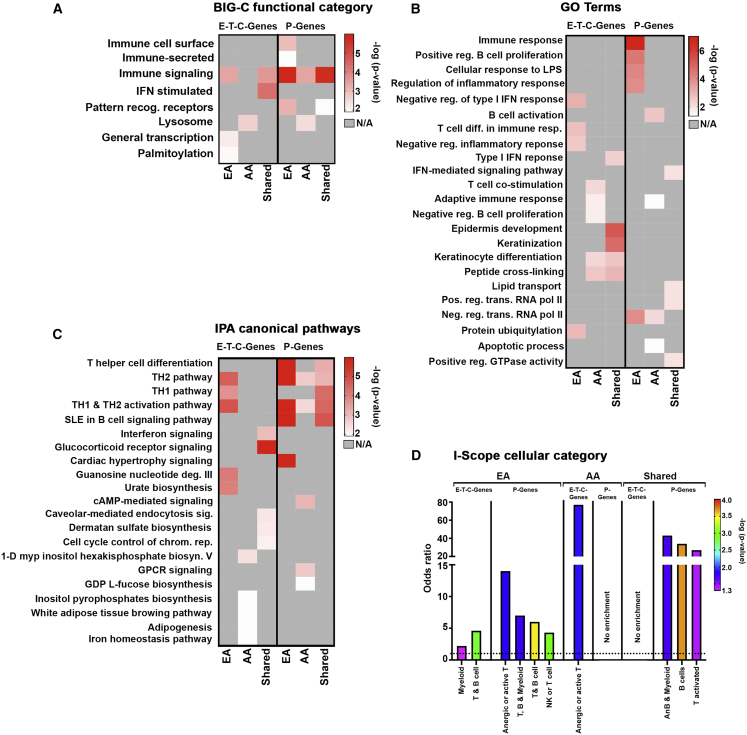
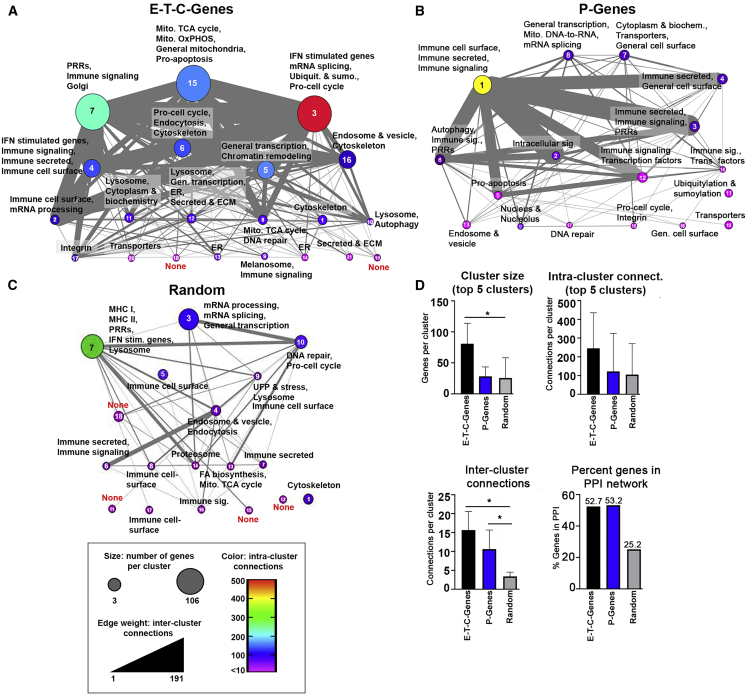
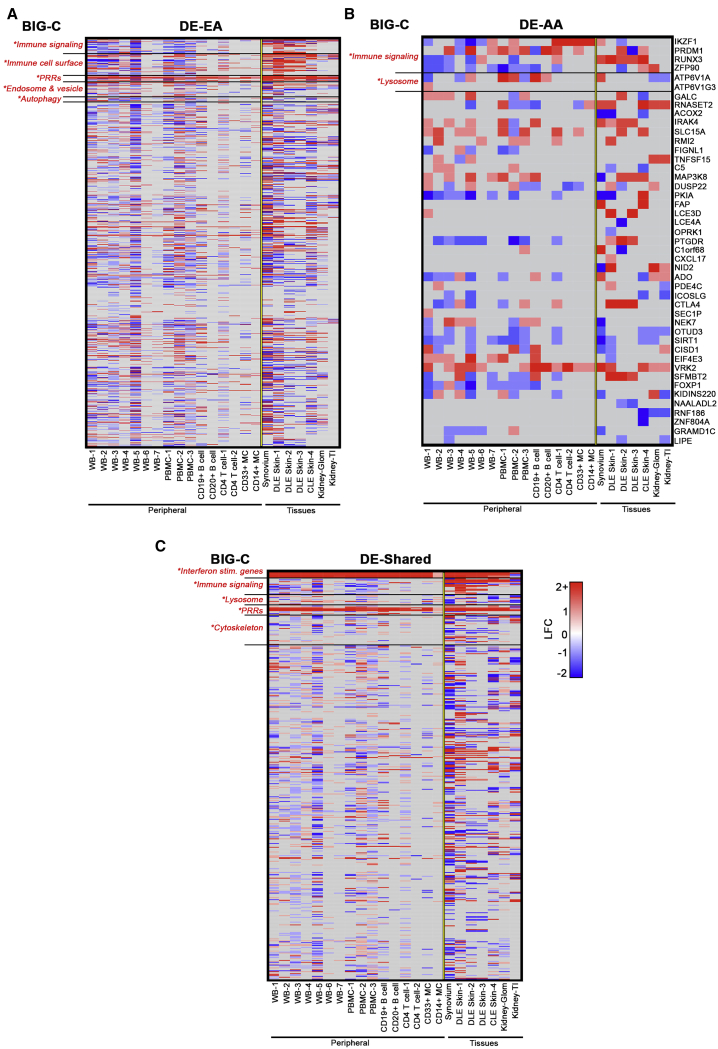


References
-
- Bentham J., Morris D.L., Graham D.S.C., Pinder C.L., Tombleson P., Behrens T.W., Martín J., Fairfax B.P., Knight J.C., Chen L. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015;47:1457–1464. - PMC - PubMed
-
- Morris D.L., Sheng Y., Zhang Y., Wang Y.F., Zhu Z., Tombleson P., Chen L., Cunninghame Graham D.S., Bentham J., Roberts A.L. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016;48:940–946. - PMC - PubMed
-
- Alarcón-Riquelme M.E., Ziegler J.T., Molineros J., Howard T.D., Moreno-Estrada A., Sánchez-Rodríguez E., Ainsworth H.C., Ortiz-Tello P., Comeau M.E., Rasmussen A. Genome-Wide Association Study in an Amerindian Ancestry Population Reveals Novel Systemic Lupus Erythematosus Risk Loci and the Role of European Admixture. Arthritis Rheumatol. 2016;68:932–943. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
