

Clinical presentation and long-term follow-up of dopamine beta hydroxylase deficiency
- PMID: 33034372
- PMCID: PMC8246878
- DOI: 10.1002/jimd.12321
Clinical presentation and long-term follow-up of dopamine beta hydroxylase deficiency
Abstract
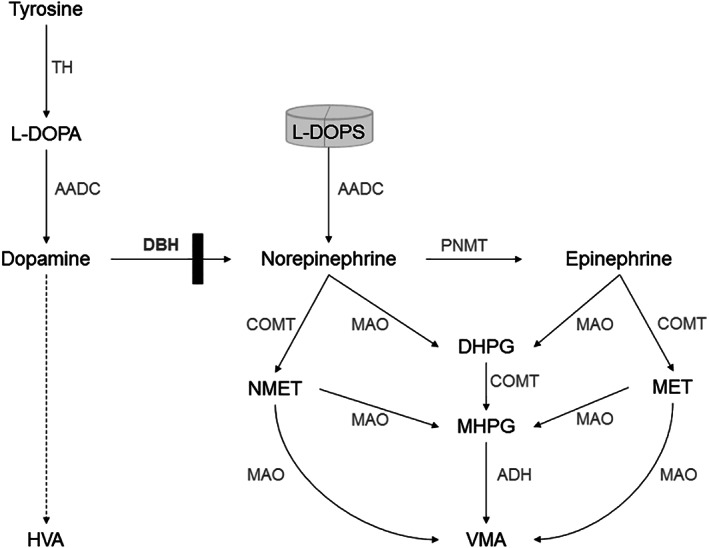
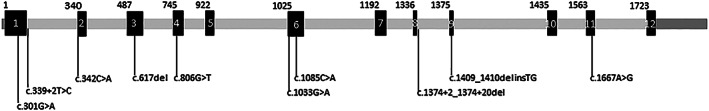
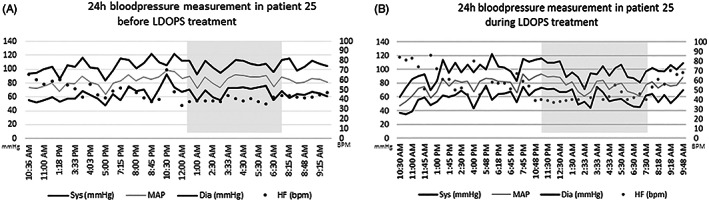
Dopamine beta hydroxylase (DBH) deficiency is an extremely rare autosomal recessive disorder with severe orthostatic hypotension, that can be treated with L-threo-3,4-dihydroxyphenylserine (L-DOPS). We aimed to summarize clinical, biochemical, and genetic data of all world-wide reported patients with DBH-deficiency, and to present detailed new data on long-term follow-up of a relatively large Dutch cohort. We retrospectively describe 10 patients from a Dutch cohort and 15 additional patients from the literature. We identified 25 patients (15 females) from 20 families. Ten patients were diagnosed in the Netherlands. Duration of follow-up of Dutch patients ranged from 1 to 21 years (median 13 years). All patients had severe orthostatic hypotension. Severely decreased or absent (nor)epinephrine, and increased dopamine plasma concentrations were found in 24/25 patients. Impaired kidney function and anemia were present in all Dutch patients, hypomagnesaemia in 5 out of 10. Clinically, all patients responded very well to L-DOPS, with marked reduction of orthostatic complaints. However, orthostatic hypotension remained present, and kidney function, anemia, and hypomagnesaemia only partially improved. Plasma norepinephrine increased and became detectable, while epinephrine remained undetectable in most patients. We confirm the core clinical characteristics of DBH-deficiency and the pathognomonic profile of catecholamines in body fluids. Impaired renal function, anemia, and hypomagnesaemia can be part of the clinical presentation. The subjective response to L-DOPS treatment is excellent and sustained, although the neurotransmitter profile in plasma does not normalize completely. Furthermore, orthostatic hypotension as well as renal function, anemia, and hypomagnesaemia improve only partially.
Keywords: L-DOPS; dopamine beta hydroxylase (DBH) deficiency; epinephrine; hypomagnesaemia; neurogenic orthostatic hypotension; neurotransmitter disorders; norepinephrine.
© 2020 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Tessa Wassenberg declares financial activities outside the submitted work: support for conference visits (travel, accommodation, and registration fees) to SSIEM 2018 Athens and SSIEM 2019 Rotterdam from Sanofi Genzyme, and speaker honorarium from PTC. Jaap Deinum, Frans J. van Ittersum, Erik‐Jan Kamsteeg, Maartje Pennings, Marcel M. Verbeek, Ron A. Wevers, Mirjam E. van Albada, Ido P. Kema, Jorie Versmissen, Ton van den Meiracker, Jacques W.M. Lenders, Leo Monnens, Michèl A. Willemsen declare that they have no conflict of interest.
Figures
References
-
- Nagatsu T. Genes for human catecholamine‐synthesizing enzymes. Neurosci Res. 1991;12(2):315‐345. - PubMed
-
- Levin EY, Levenberg B, Kaufman S. The enzymatic conversion of 3,4‐dihydroxyphenylethylamine to norepinephrine. J Biol Chem. 1960;235:2080‐2086. - PubMed
-
- Robertson D, Garland EM. Dopamine beta‐hydroxylase deficiency. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews. Seattle, WA: University of Washington; 1993. - PubMed
-
- Robertson D, Goldberg MR, Onrot J, et al. Isolated failure of autonomic noradrenergic neurotransmission. Evidence for impaired beta‐hydroxylation of dopamine. N Engl J Med. 1986;314(23):1494‐1497. - PubMed
-
- Man in't Veld AJ, Boomsma F, Moleman P, Schalekamp MA. Congenital dopamine‐beta‐hydroxylase deficiency. A novel orthostatic syndrome. Lancet. 1987;1(8526):183‐188. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
