

Using quantum chemistry to estimate chemical shifts in biomolecules
- PMID: 33035752
- PMCID: PMC7686263
- DOI: 10.1016/j.bpc.2020.106476
Using quantum chemistry to estimate chemical shifts in biomolecules
Abstract
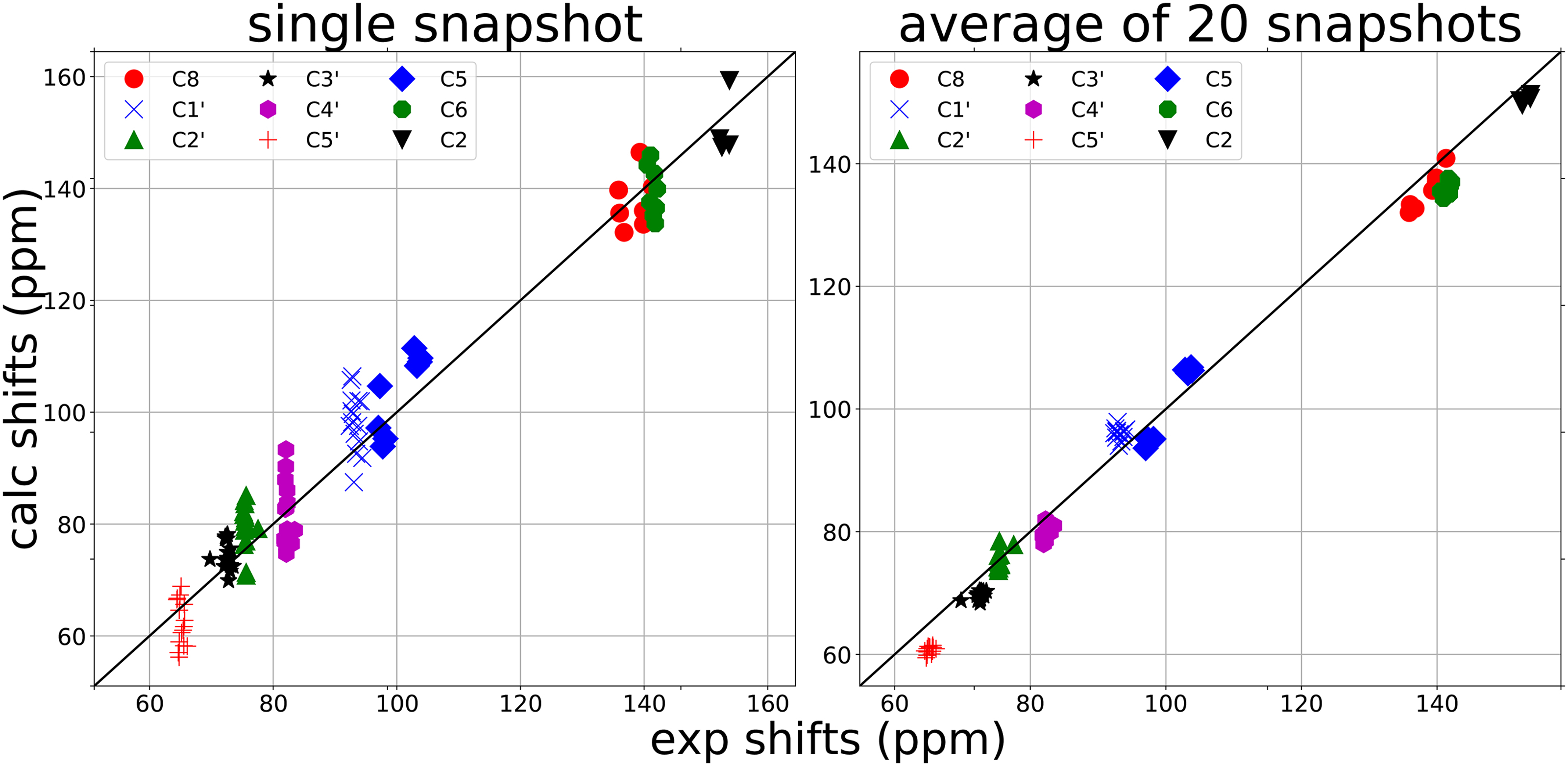
An automated fragmentation quantum mechanics/molecular mechanics approach (AFNMR) has shown promising results in chemical shift calculations for biomolecules. Sample results for ubiquitin, and an RNA hairpin and helix are presented, and used to recent directions in quantum calculations. Trends in chemical shift are stable with regards to change in density functional or basis sets, and the use of the small "pcSseg-0" basis, which was optimized for chemical shift prediction [1], opens the way to more extensive conformational averaging, which can often be necessary, even for fairly well-defined structures.
Keywords: Chemical shift NMR conformational averaging.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Figures
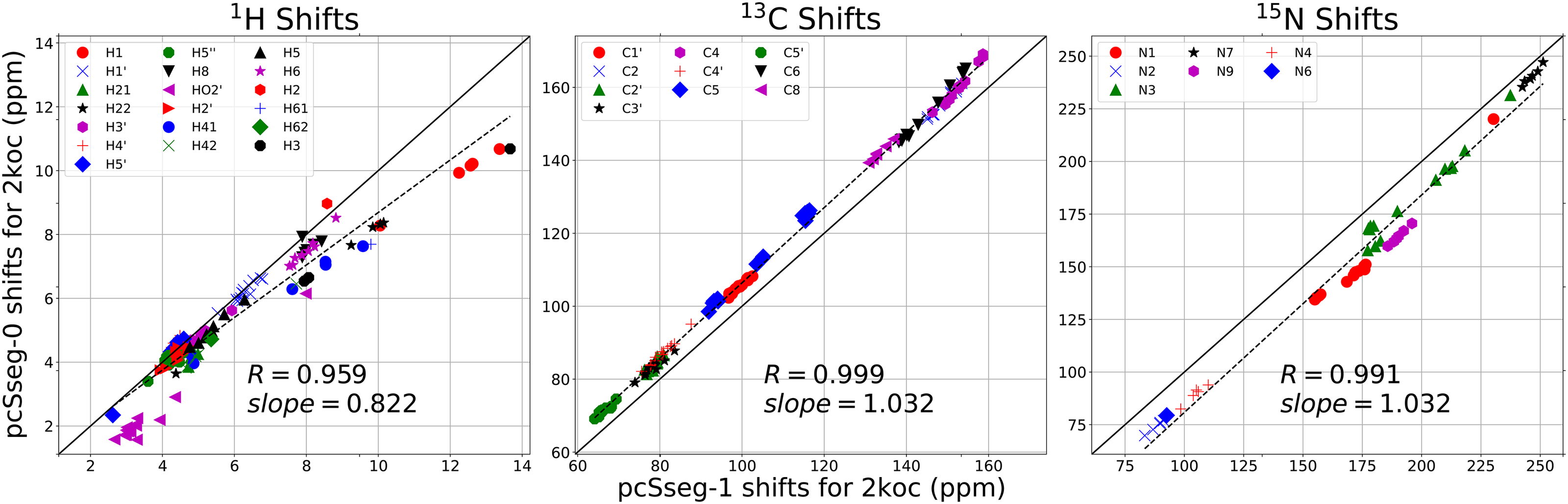
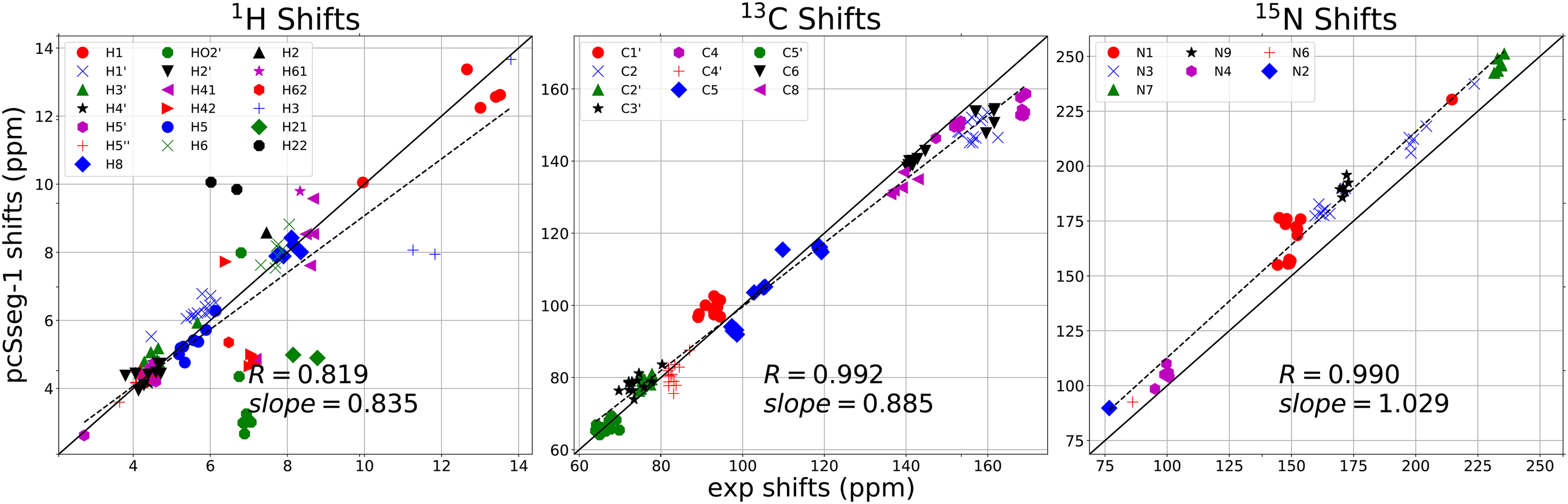
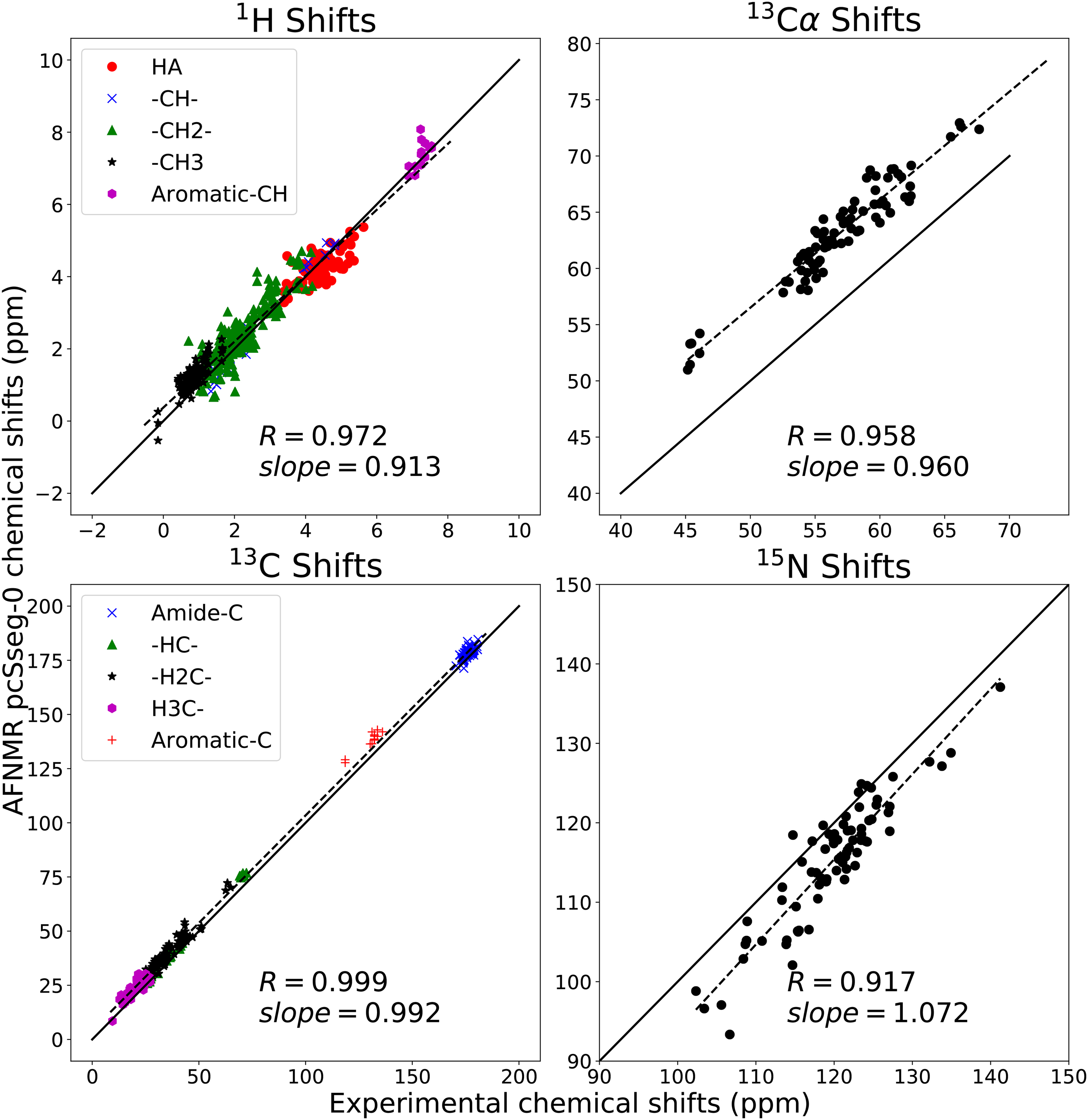
References
-
- Jensen F, Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding, J. Chem. Theory Comput 11 (2015) 132–138. - PubMed
-
- Buckingham A, Schaefer T, Schneider W, Solvent effects in nuclear magnetic resonance spectra, J. Chem. Phys 32 (1960) 1227–1233.
-
- Sitkoff D, Case D, Density functional calculations of proton chemical shifts in model peptides, J. Am. Chem. Soc 119 (1997) 12262–12273.
-
- Sitkoff D, Case D, Theories of chemical shift anisotropies in proteins and nucleic acids, Prog. NMR Spectr 32 (1998) 165–190.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
