

Targeting the GCK pathway: a novel and selective therapeutic strategy against RAS-mutated multiple myeloma
- PMID: 33036022
- PMCID: PMC8020269
- DOI: 10.1182/blood.2020006334
Targeting the GCK pathway: a novel and selective therapeutic strategy against RAS-mutated multiple myeloma
Abstract
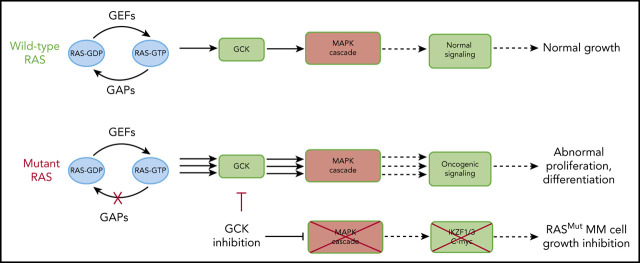
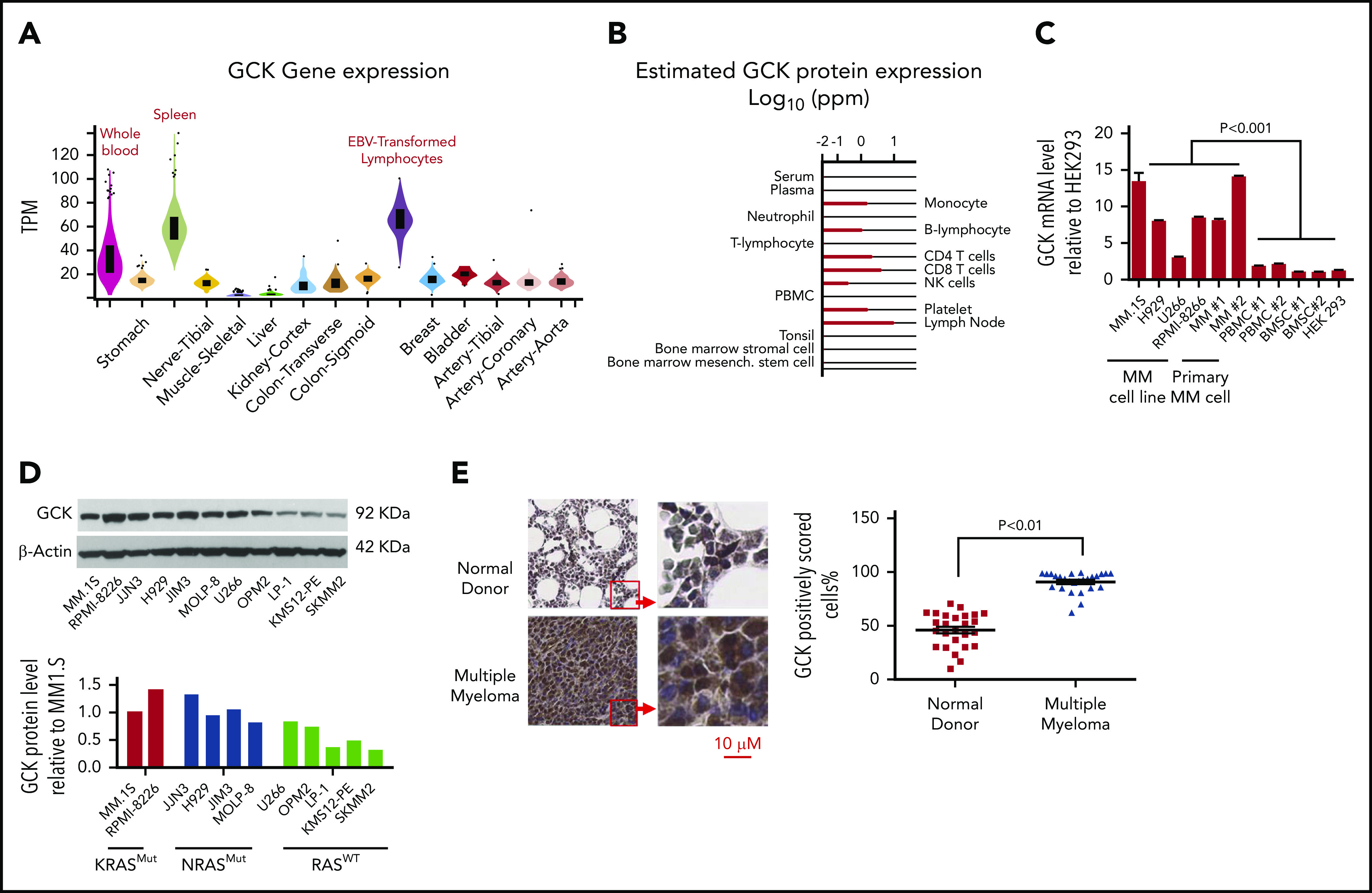
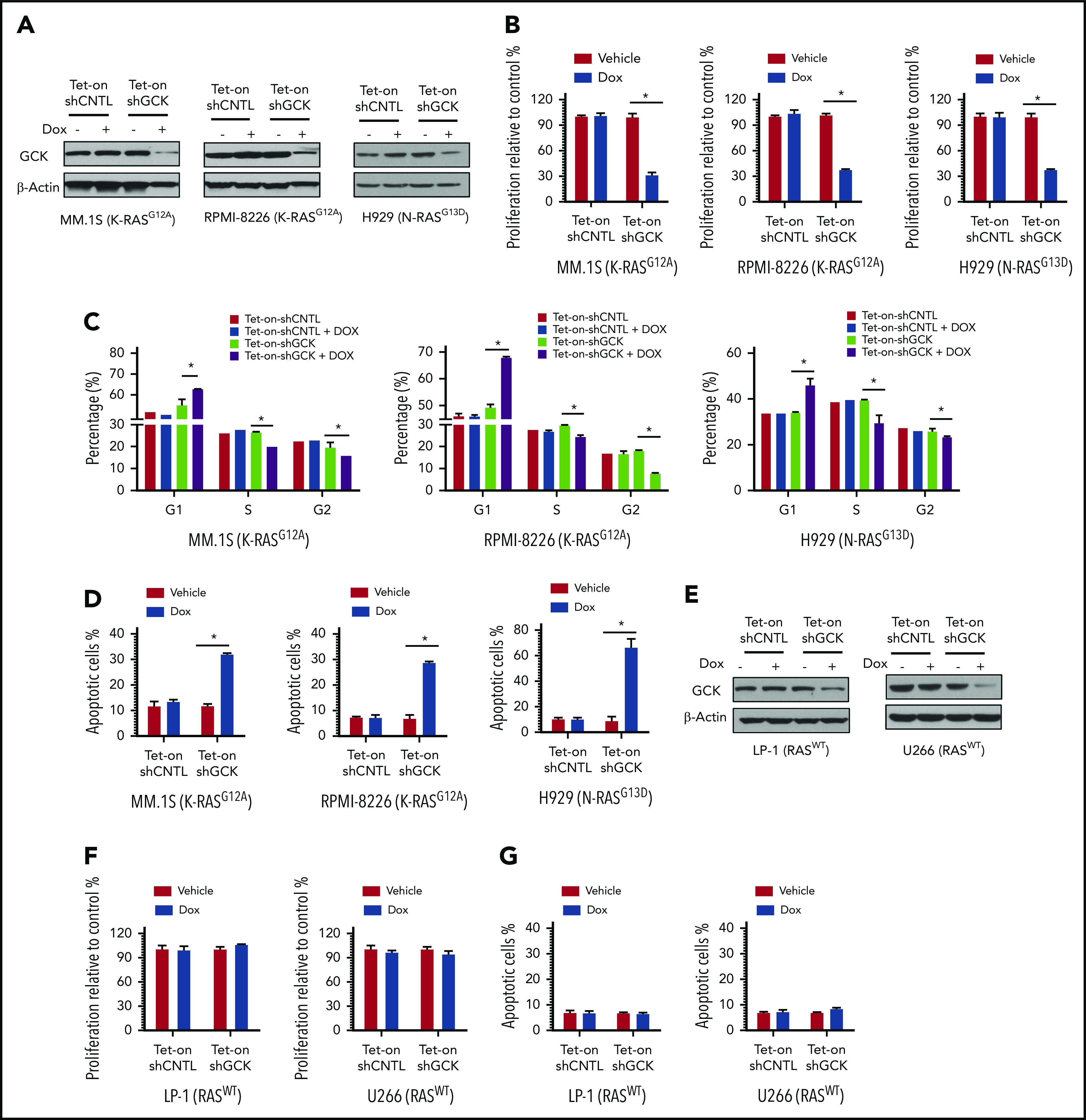
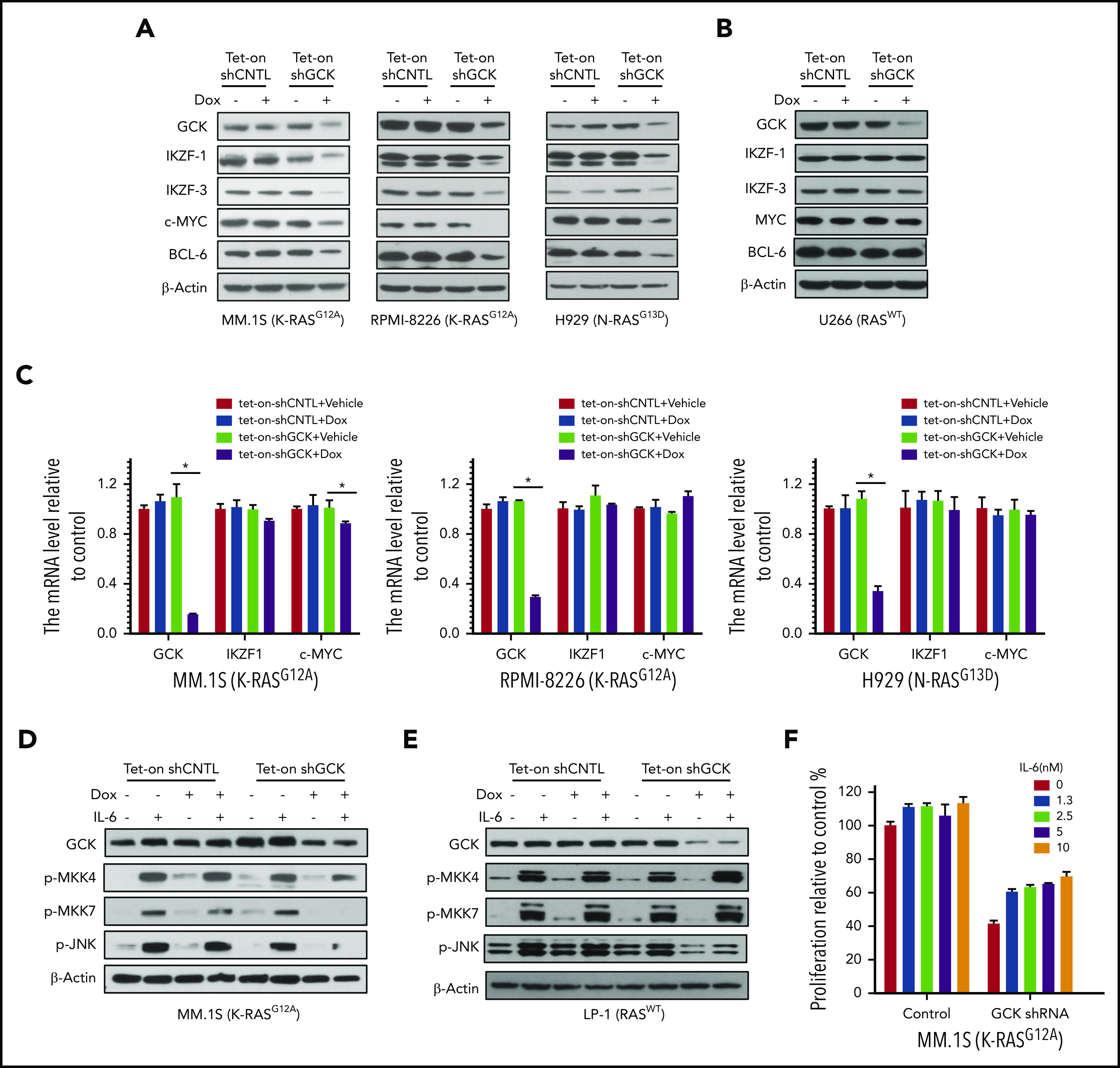
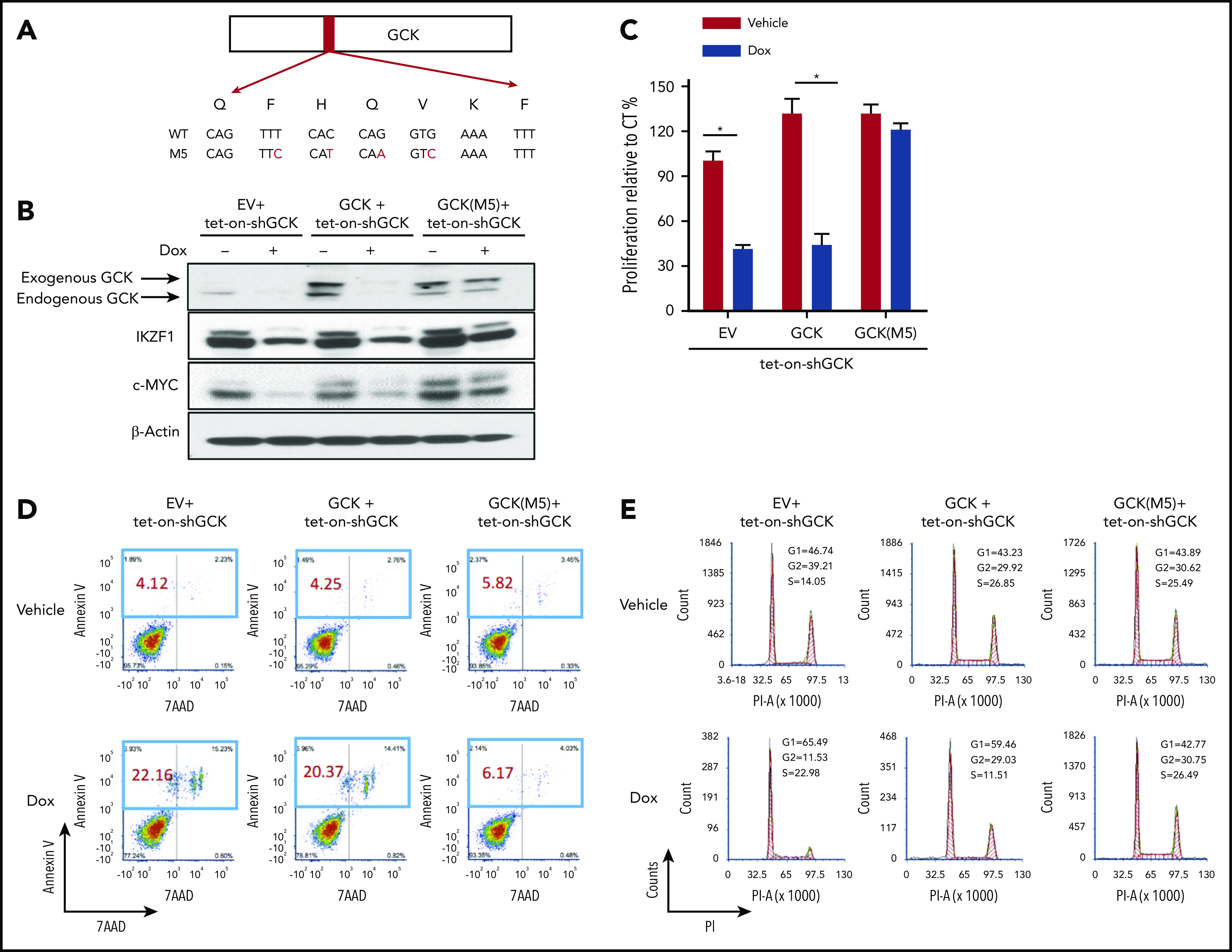
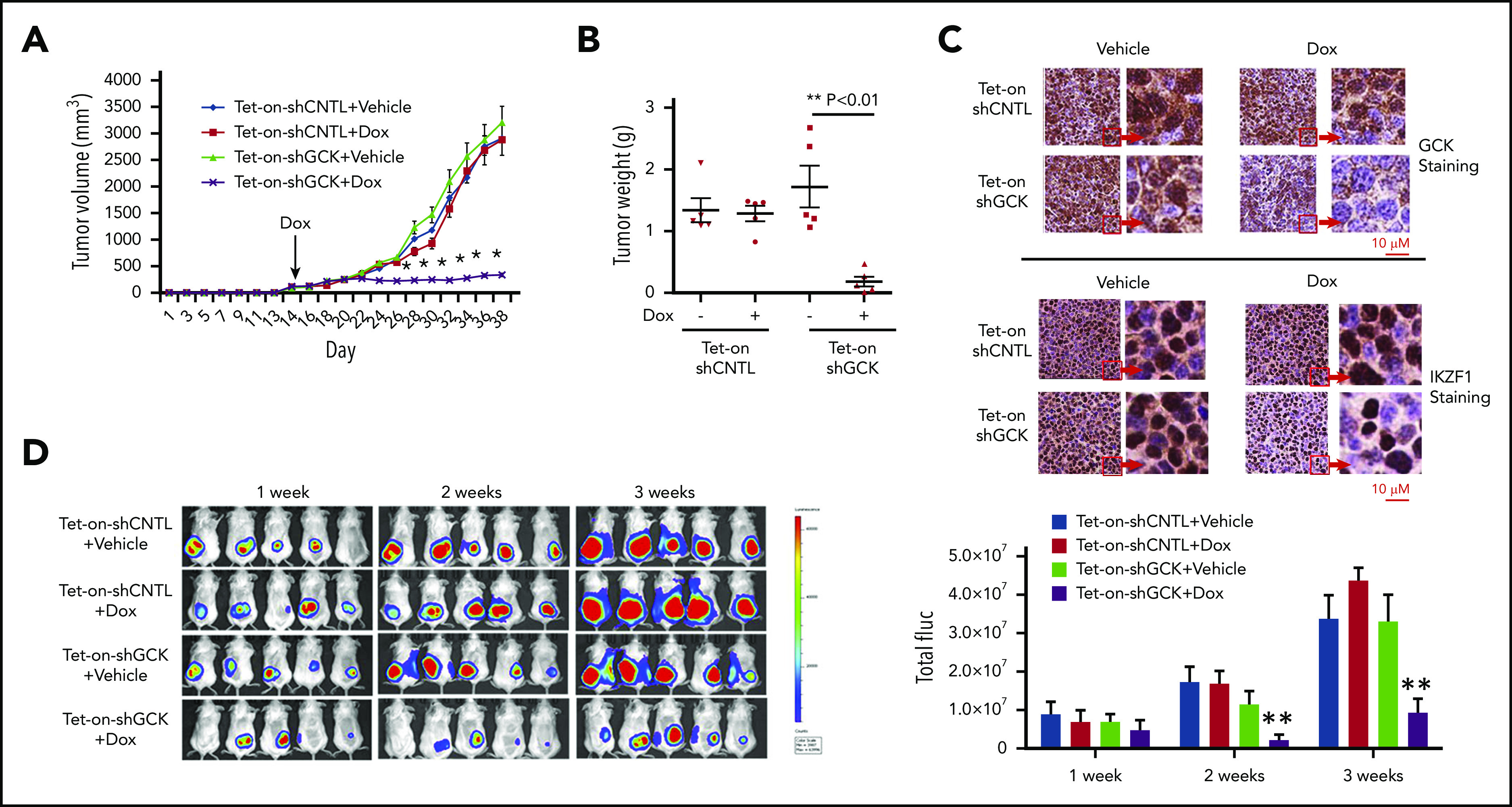
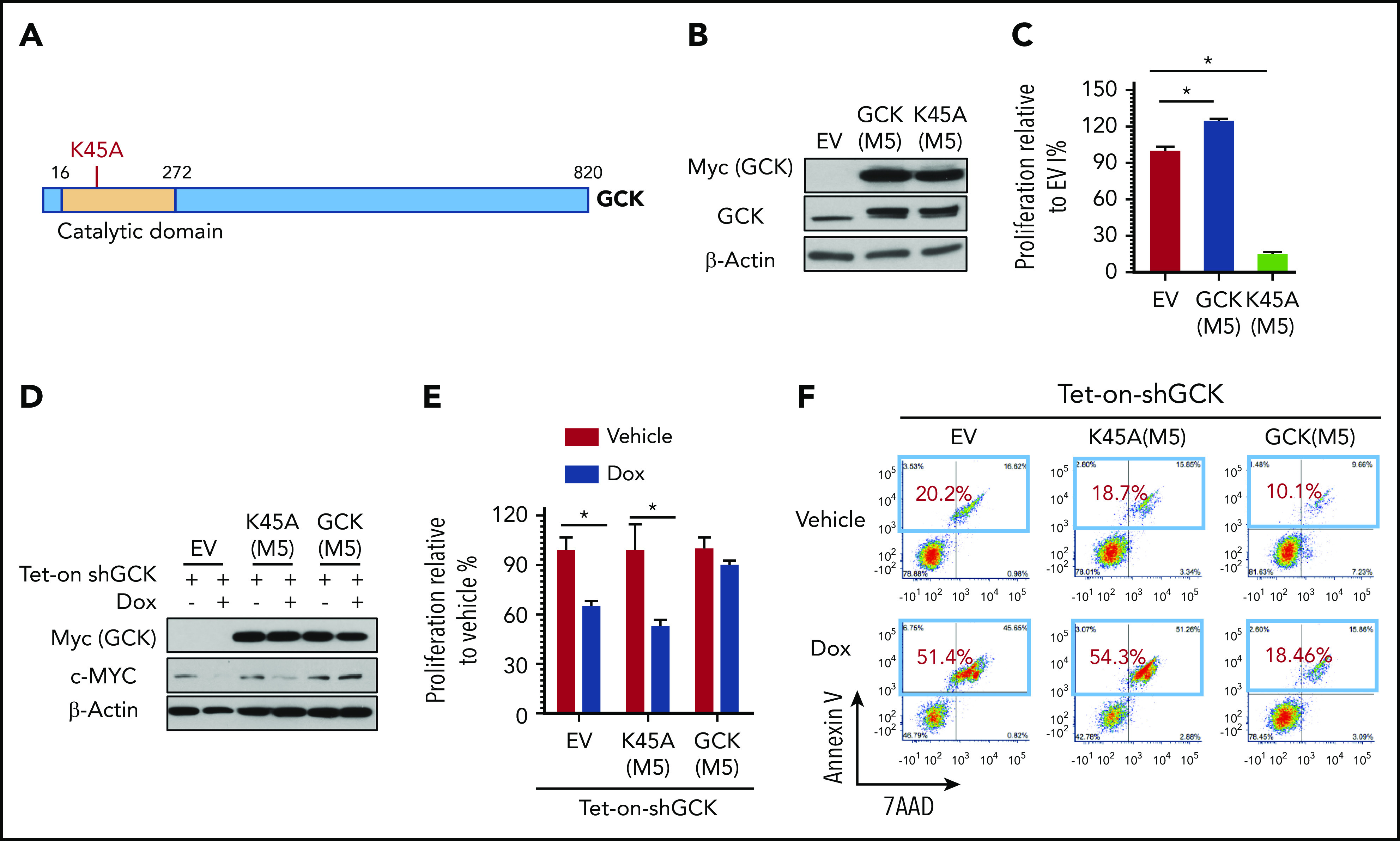
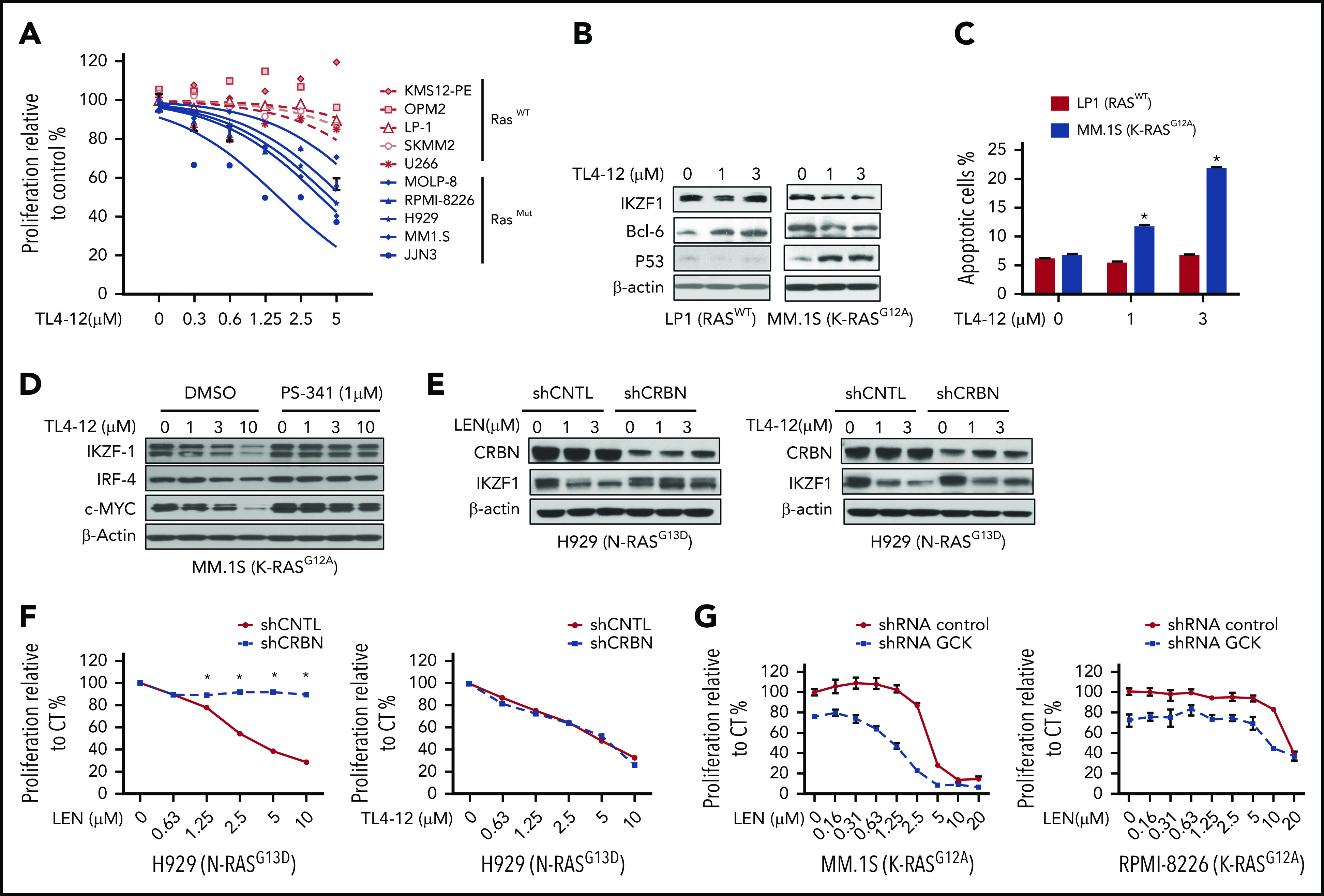
In multiple myeloma (MM), frequent mutations of NRAS, KRAS, or BRAF are found in up to 50% of newly diagnosed patients. The majority of the NRAS, KRAS, and BRAF mutations occur in hotspots causing constitutive activation of the corresponding proteins. Thus, targeting RAS mutation in MM will increase therapeutic efficiency and potentially overcome drug resistance. We identified germinal center kinase (GCK) as a novel therapeutic target in MM with RAS mutation. GCK knockdown (KD) in MM cells demonstrated in vitro and in vivo that silencing of GCK induces MM cell growth inhibition, associated with blocked MKK4/7-JNK phosphorylation and impaired degradation of IKZF1/3, BCL-6, and c-MYC. These effects were rescued by overexpression of a short hairpin RNA (shRNA)-resistant GCK, thereby excluding the potential off-target effects of GCK KD. In contrast, overexpression of shRNA-resistant GCK kinase-dead mutant (K45A) inhibited MM cell proliferation and failed to rescue the effects of GCK KD on MM growth inhibition, indicating that GCK kinase activity is critical for regulating MM cell proliferation and survival. Importantly, the higher sensitivity to GCK KD in RASMut cells suggests that targeting GCK is effective in MM, which harbors RAS mutations. In accordance with the effects of GCK KD, the GCK inhibitor TL4-12 dose-dependently downregulated IKZF1 and BCL-6 and led to MM cell proliferation inhibition accompanied by induction of apoptosis. Here, our data identify GCK as a novel target in RASMut MM cells, providing a rationale to treat RAS mutations in MM. Furthermore, GCK inhibitors might represent an alternative therapy to overcome immunomodulatory drug resistance in MM.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: S. Lentzsch reports Caelum Biosciences equity ownership and membership on Caelum Bioscience's board of directors or advisory committees; consultancy for Janssen, Takeda, GSK, Antengene, Adaptive and Sorrento; and received research funding from Karyopharm and Sanofi. M.Y.M. reports receiving research funding from Ossium Health, Inc and consultancy for Ossium Health. C.M. is a full-time employee of Sanofi. The remaining authors declare no competing financial interests.
Figures
References
-
- Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3(1):17046. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30. - PubMed
-
- Moreau P. How I treat myeloma with new agents. Blood. 2017;130(13):1507-1513. - PubMed
-
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459-465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
