

Fundamental Difficulties Prevent the Reconstruction of the Deep Phylogeny of Viruses
- PMID: 33036160
- PMCID: PMC7600955
- DOI: 10.3390/v12101130
Fundamental Difficulties Prevent the Reconstruction of the Deep Phylogeny of Viruses
Abstract
The extension of virology beyond its traditional medical, veterinary, or agricultural applications, now called environmental virology, has shown that viruses are both the most numerous and diverse biological entities on Earth. In particular, virus isolations from unicellular eukaryotic hosts (heterotrophic and photosynthetic protozoans) revealed numerous viral types previously unexpected in terms of virion structure, gene content, or mode of replication. Complemented by large-scale metagenomic analyses, these discoveries have rekindled interest in the enigma of the origin of viruses, for which a description encompassing all their diversity remains not available. Several laboratories have repeatedly tackled the deep reconstruction of the evolutionary history of viruses, using various methods of molecular phylogeny applied to the few shared "core" genes detected in certain virus groups (e.g., the Nucleocytoviricota). Beyond the practical difficulties of establishing reliable homology relationships from extremely divergent sequences, I present here conceptual arguments highlighting several fundamental limitations plaguing the reconstruction of the deep evolutionary history of viruses, and even more the identification of their unique or multiple origin(s). These arguments also underline the risk of establishing premature high level viral taxonomic classifications. Those limitations are direct consequences of the random mechanisms governing the reductive/retrogressive evolution of all obligate intracellular parasites.
Keywords: Bamfordvirae; Nucleocytoviricota; Varidnaviria; obligate intracellular parasites; origin of viruses; phylogenetic reconstruction; reductive evolution.
Conflict of interest statement
The author declares no conflict of interest.
Figures
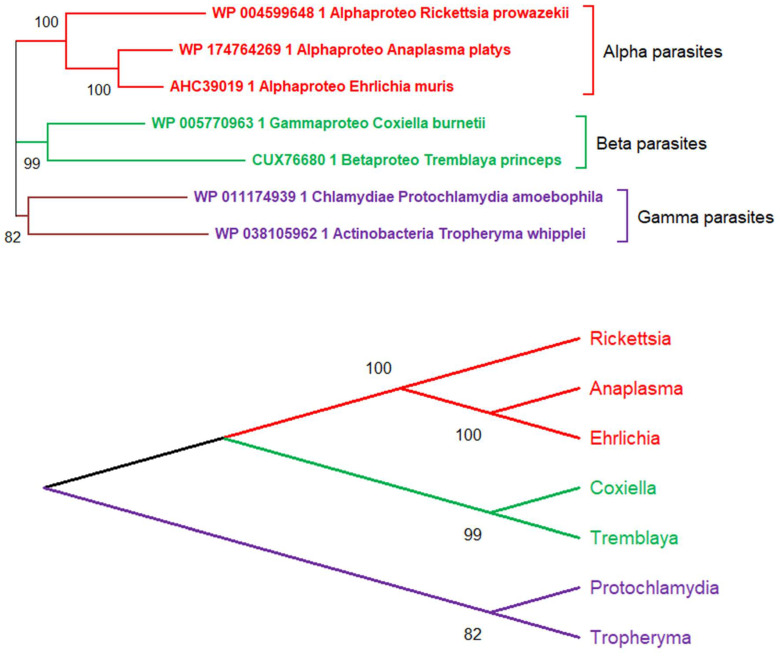
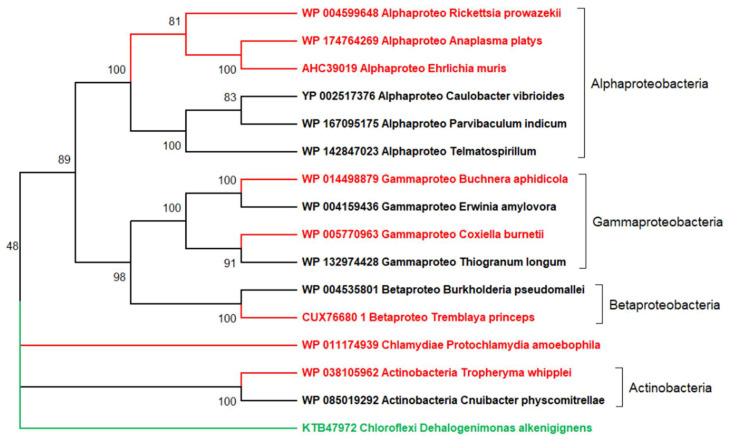
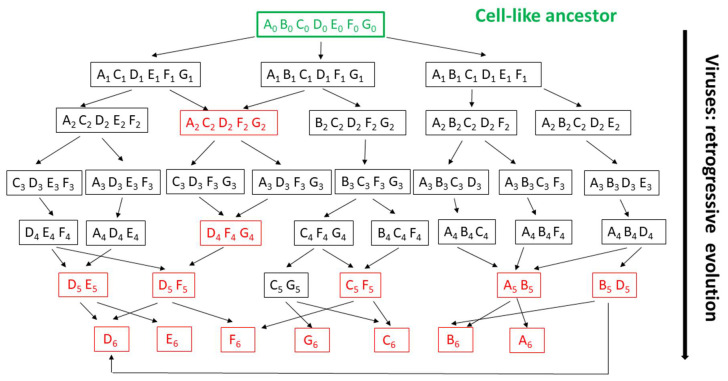
References
-
- Ivanovski D. In: Über die Mosaikkrankheit der Tabakspflanze. Johnson J., translator. American Phytopathological Society; St. Paul, MN, USA: 1892. pp. 27–30. Izv. Imp. Akad. Nauk. 1892; 35, 67. Phytopathological classics.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
