

Presentation and management of N-acetylglutamate synthase deficiency: a review of the literature
- PMID: 33036647
- PMCID: PMC7545900
- DOI: 10.1186/s13023-020-01560-z
Presentation and management of N-acetylglutamate synthase deficiency: a review of the literature
Abstract
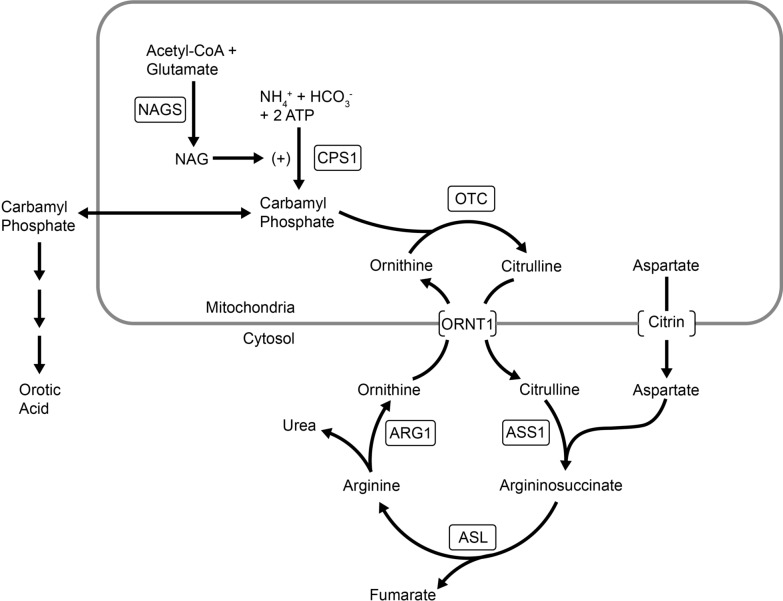
Background: N-Acetylglutamate synthase (NAGS) deficiency is an extremely rare autosomal recessive metabolic disorder affecting the urea cycle, leading to episodes of hyperammonemia which can cause significant morbidity and mortality. Since its recognition in 1981, NAGS deficiency has been treated with carbamylglutamate with or without other measures (nutritional, ammonia scavengers, dialytic, etc.). We conducted a systematic literature review of NAGS deficiency to summarize current knowledge around presentation and management.
Methods: Case reports and case series were identified using the Medline database, as well as references from other articles and a general internet search. Clinical data related to presentation and management were abstracted by two reviewers.
Results: In total, 98 cases of NAGS deficiency from 79 families, in 48 articles or abstracts were identified. Of these, 1 was diagnosed prenatally, 57 were neonatal cases, 34 were post-neonatal, and 6 did not specify age at presentation or were asymptomatic at diagnosis. Twenty-one cases had relevant family history. We summarize triggers of hyperammonemic episodes, diagnosis, clinical signs and symptoms, and management strategies. DNA testing is the preferred method of diagnosis, although therapeutic trials to assess response of ammonia levels to carbamylglutamate may also be helpful. Management usually consists of treatment with carbamylglutamate, although the reported maintenance dose varied across case reports. Protein restriction was sometimes used in conjunction with carbamylglutamate. Supplementation with citrulline, arginine, and sodium benzoate also were reported.
Conclusions: Presentation of NAGS deficiency varies by age and symptoms. In addition, both diagnosis and management have evolved over time and vary across clinics. Prompt recognition and appropriate treatment of NAGS deficiency with carbamylglutamate may improve outcomes of affected individuals. Further research is needed to assess the roles of protein restriction and supplements in the treatment of NAGS deficiency, especially during times of illness or lack of access to carbamylglutamate.
Keywords: Carbaglu; Carbamylglutamate; Hyperammonemia; Inherited metabolic disorder; N-Acetylglutamate synthase; N-Acetylglutamate synthase deficiency; NAGS; Urea cycle.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea cycle disorders overview. 2003 Apr 29 (Updated 2017 Jun 22). In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. https://www.ncbi.nlm.nih.gov/books/NBK1217/. - PubMed
-
- Bachmann C, Krahenbuhl S, Colombo JP, Schubiger G, Jaggi KH, Tonz O. N-Acetylglutamate synthetase deficiency: a disorder of ammonia detoxication. N Engl J Med. 1981;304(9):543. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
