

Leptin modulates pancreatic β-cell membrane potential through Src kinase-mediated phosphorylation of NMDA receptors
- PMID: 33037073
- PMCID: PMC7863909
- DOI: 10.1074/jbc.RA120.015489
Leptin modulates pancreatic β-cell membrane potential through Src kinase-mediated phosphorylation of NMDA receptors
Abstract
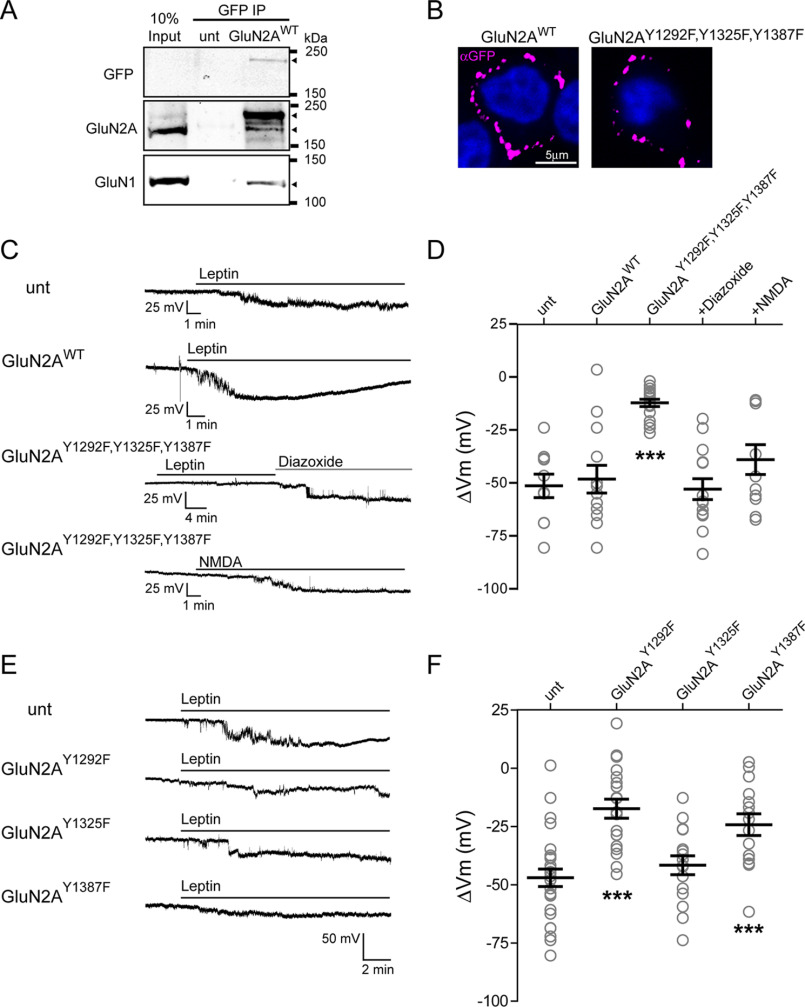
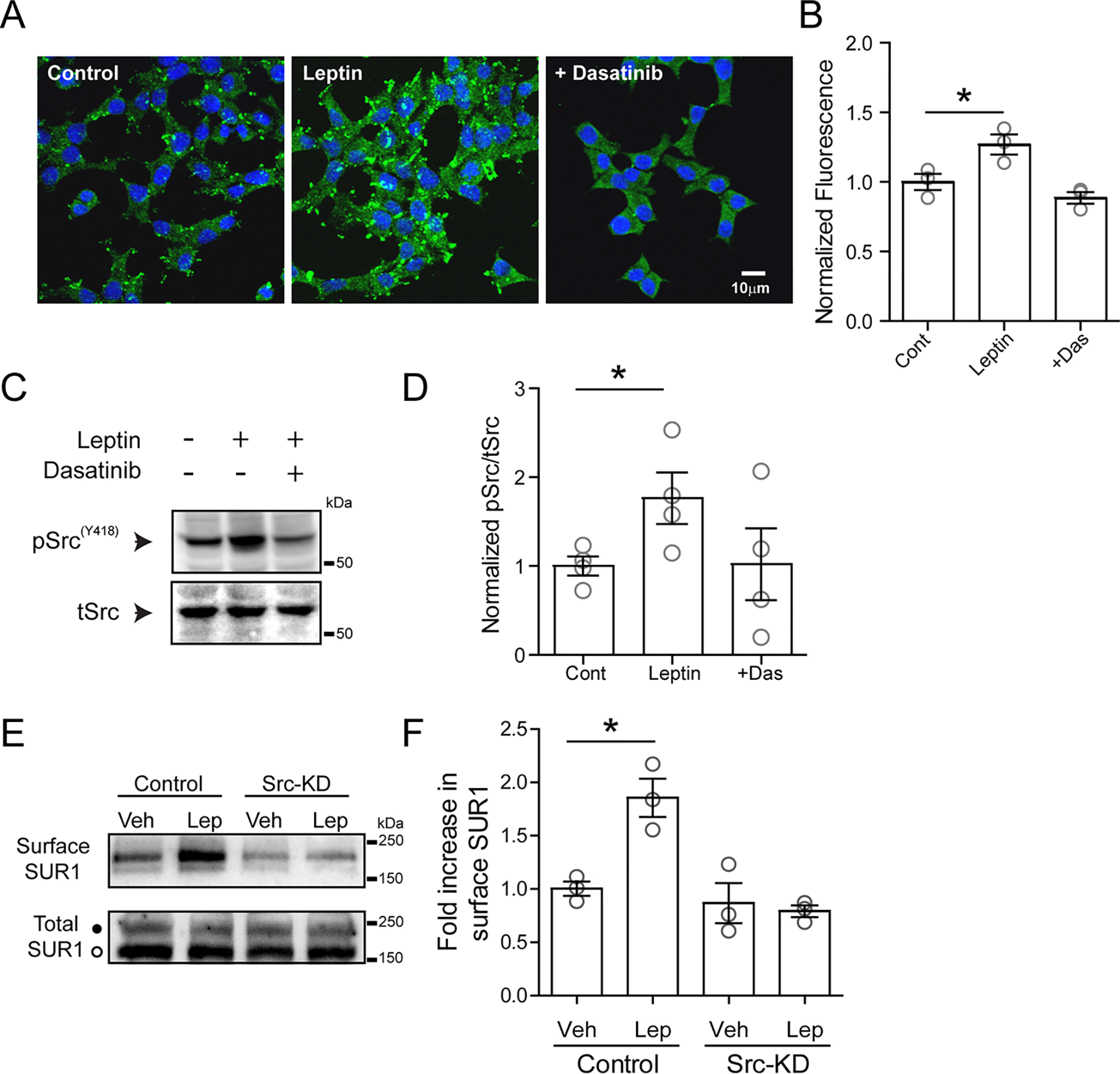
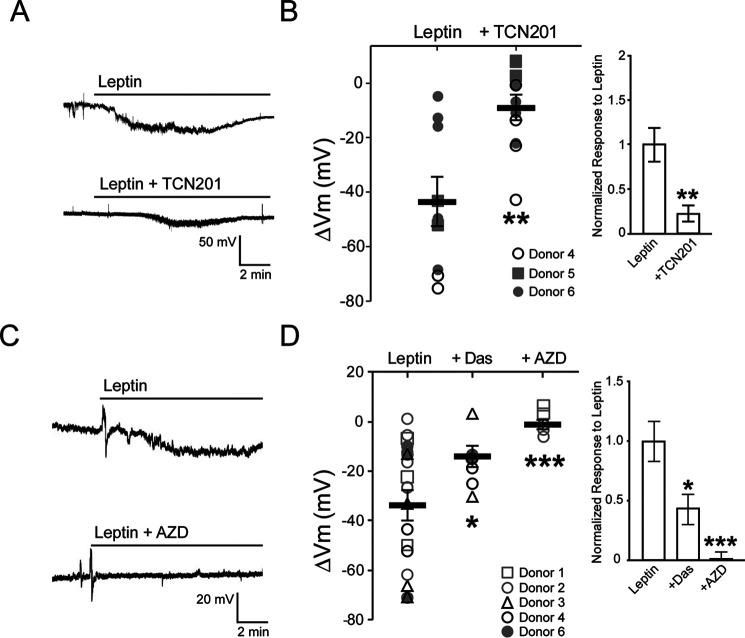
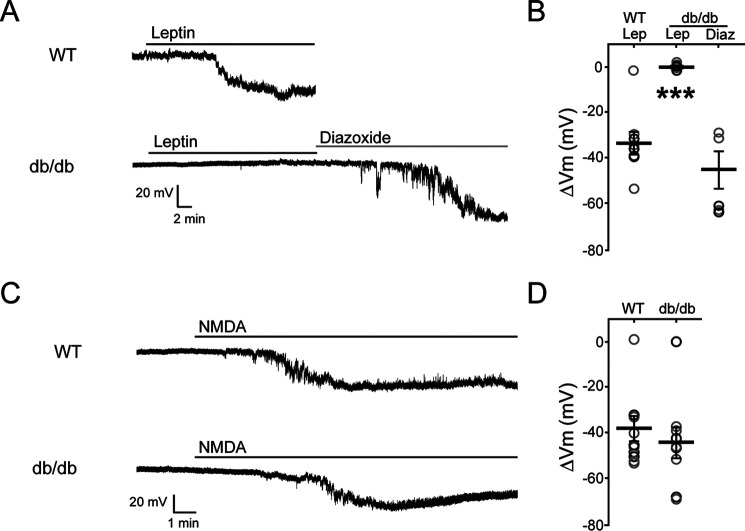
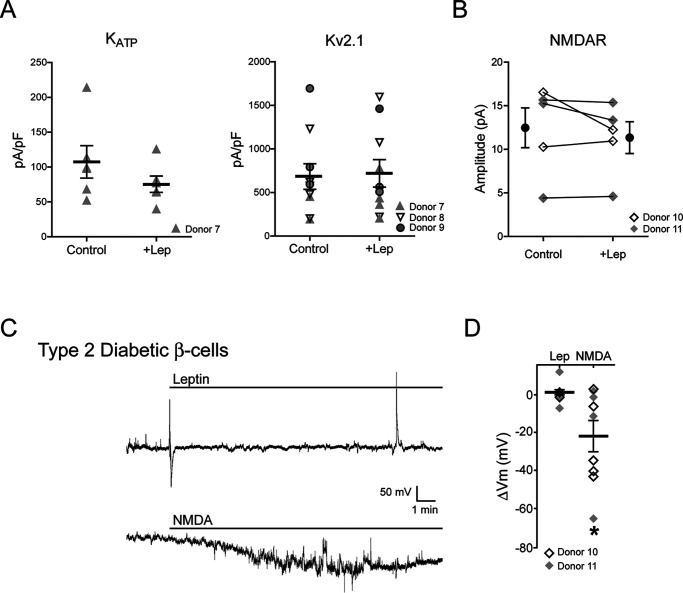
The adipocyte-derived hormone leptin increases trafficking of KATP and Kv2.1 channels to the pancreatic β-cell surface, resulting in membrane hyperpolarization and suppression of insulin secretion. We have previously shown that this effect of leptin is mediated by the NMDA subtype of glutamate receptors (NMDARs). It does so by potentiating NMDAR activity, thus enhancing Ca2+ influx and the ensuing downstream signaling events that drive channel trafficking to the cell surface. However, the molecular mechanism by which leptin potentiates NMDARs in β-cells remains unknown. Here, we report that leptin augments NMDAR function via Src kinase-mediated phosphorylation of the GluN2A subunit. Leptin-induced membrane hyperpolarization diminished upon pharmacological inhibition of GluN2A but not GluN2B, indicating involvement of GluN2A-containing NMDARs. GluN2A harbors tyrosine residues that, when phosphorylated by Src family kinases, potentiate NMDAR activity. We found that leptin increases phosphorylation of Tyr-418 in Src, an indicator of kinase activation. Pharmacological inhibition of Src or overexpression of a kinase-dead Src mutant prevented the effect of leptin, whereas a Src kinase activator peptide mimicked it. Using mutant GluN2A overexpression, we show that Tyr-1292 and Tyr-1387 but not Tyr-1325 are responsible for the effect of leptin. Importantly, β-cells from db/db mice, a type 2 diabetes mouse model lacking functional leptin receptors, or from obese diabetic human donors failed to respond to leptin but hyperpolarized in response to NMDA. Our study reveals a signaling pathway wherein leptin modulates NMDARs via Src to regulate β-cell excitability and suggests NMDARs as a potential target to overcome leptin resistance.
Keywords: GluN2A; KATP channel; Src; Src kinase; beta cell (B-cell); glutamate receptor; phosphorylation; type 2 diabetes; β-cells.
© 2020 Cochrane et al.
Conflict of interest statement
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Kulkarni, R. N., Wang, Z. L., Wang, R. M., Hurley, J. D., Smith, D. M., Ghatei, M. A., Withers, D. J., Gardiner, J. V., Bailey, C. J., and Bloom, S. R. (1997) Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Invest. 100, 2729–2736 10.1172/JCI119818 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
