

Accommodating individual travel history and unsampled diversity in Bayesian phylogeographic inference of SARS-CoV-2
- PMID: 33037213
- PMCID: PMC7547076
- DOI: 10.1038/s41467-020-18877-9
Accommodating individual travel history and unsampled diversity in Bayesian phylogeographic inference of SARS-CoV-2
Abstract
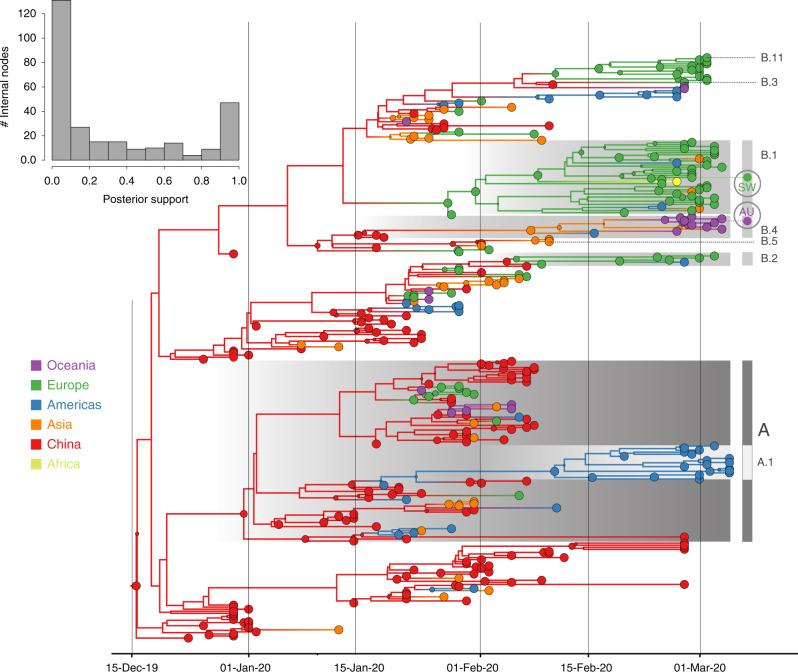
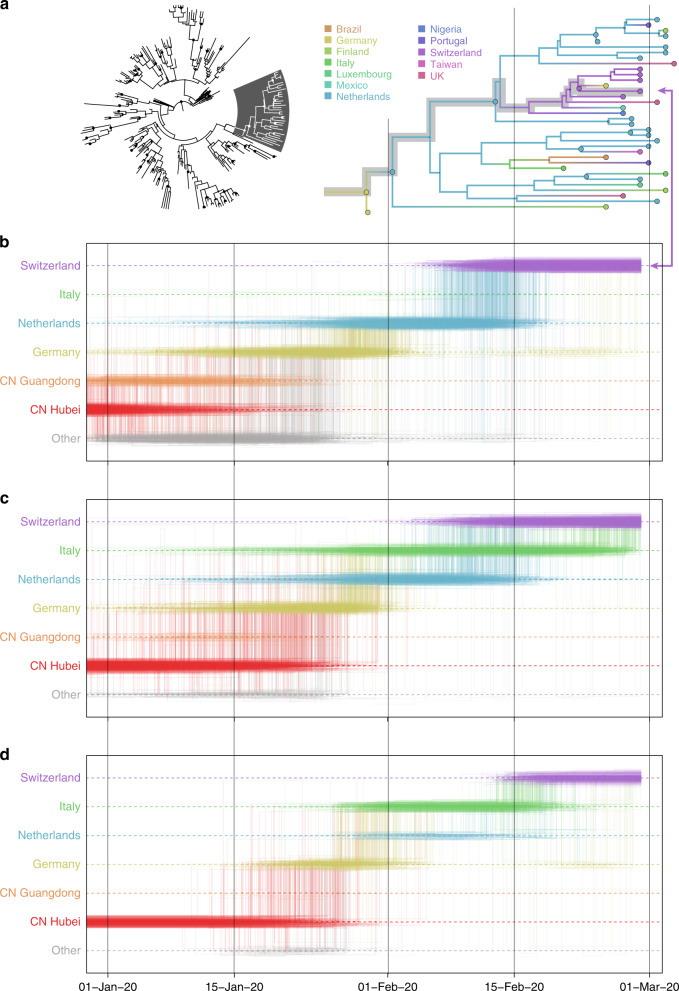
Spatiotemporal bias in genome sampling can severely confound discrete trait phylogeographic inference. This has impeded our ability to accurately track the spread of SARS-CoV-2, the virus responsible for the COVID-19 pandemic, despite the availability of unprecedented numbers of SARS-CoV-2 genomes. Here, we present an approach to integrate individual travel history data in Bayesian phylogeographic inference and apply it to the early spread of SARS-CoV-2. We demonstrate that including travel history data yields i) more realistic hypotheses of virus spread and ii) higher posterior predictive accuracy compared to including only sampling location. We further explore methods to ameliorate the impact of sampling bias by augmenting the phylogeographic analysis with lineages from undersampled locations. Our reconstructions reinforce specific transmission hypotheses suggested by the inclusion of travel history data, but also suggest alternative routes of virus migration that are plausible within the epidemiological context but are not apparent with current sampling efforts.
Conflict of interest statement
The authors declare no competing interests.
Figures





Update of
-
Accommodating individual travel history, global mobility, and unsampled diversity in phylogeography: a SARS-CoV-2 case study.bioRxiv [Preprint]. 2020 Jun 23:2020.06.22.165464. doi: 10.1101/2020.06.22.165464. bioRxiv. 2020. Update in: Nat Commun. 2020 Oct 9;11(1):5110. doi: 10.1038/s41467-020-18877-9. PMID: 32596695 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
