

Proteomic and transcriptomic profiling of aerial organ development in Arabidopsis
- PMID: 33037224
- PMCID: PMC7547660
- DOI: 10.1038/s41597-020-00678-w
Proteomic and transcriptomic profiling of aerial organ development in Arabidopsis
Abstract
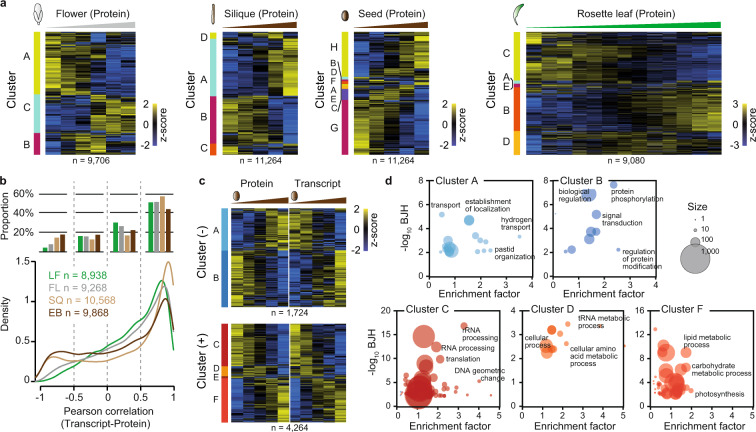
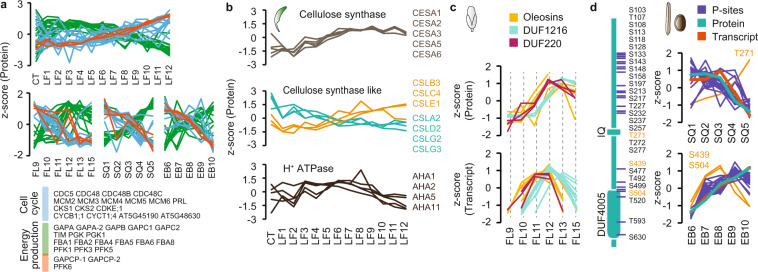
Plant growth and development are regulated by a tightly controlled interplay between cell division, cell expansion and cell differentiation during the entire plant life cycle from seed germination to maturity and seed propagation. To explore some of the underlying molecular mechanisms in more detail, we selected different aerial tissue types of the model plant Arabidopsis thaliana, namely rosette leaf, flower and silique/seed and performed proteomic, phosphoproteomic and transcriptomic analyses of sequential growth stages using tandem mass tag-based mass spectrometry and RNA sequencing. With this exploratory multi-omics dataset, development dynamics of photosynthetic tissues can be investigated from different angles. As expected, we found progressive global expression changes between growth stages for all three omics types and often but not always corresponding expression patterns for individual genes on transcript, protein and phosphorylation site level. The biggest difference between proteomic- and transcriptomic-based expression information could be observed for seed samples. Proteomic and transcriptomic data is available via ProteomeXchange and ArrayExpress with the respective identifiers PXD018814 and E-MTAB-7978.
Conflict of interest statement
M.W. and B.K. are founders and shareholders of OmicScouts GmbH and msAId GmbH. They have no operational role in the companies. M.F. is founder, shareholder and the CEO of msAId GmbH. The remaining authors declare no competing interests.
Figures



Comment on
-
Mass-spectrometry-based draft of the Arabidopsis proteome.Nature. 2020 Mar;579(7799):409-414. doi: 10.1038/s41586-020-2094-2. Epub 2020 Mar 11. Nature. 2020. PMID: 32188942
References
Publication types
MeSH terms
Substances
Grants and funding
- SFB924/Deutsche Forschungsgemeinschaft (German Research Foundation)/International
- SFB924/Deutsche Forschungsgemeinschaft (German Research Foundation)/International
- SFB924/Deutsche Forschungsgemeinschaft (German Research Foundation)/International
- SFB924/Deutsche Forschungsgemeinschaft (German Research Foundation)/International
- SFB924/Deutsche Forschungsgemeinschaft (German Research Foundation)/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
