

SUV39H1 regulates the progression of MLL-AF9-induced acute myeloid leukemia
- PMID: 33037410
- PMCID: PMC7728597
- DOI: 10.1038/s41388-020-01495-6
SUV39H1 regulates the progression of MLL-AF9-induced acute myeloid leukemia
Abstract
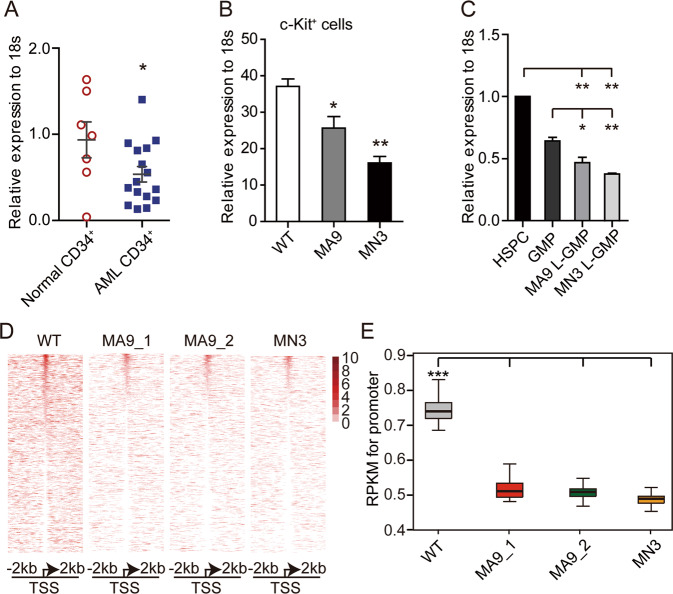
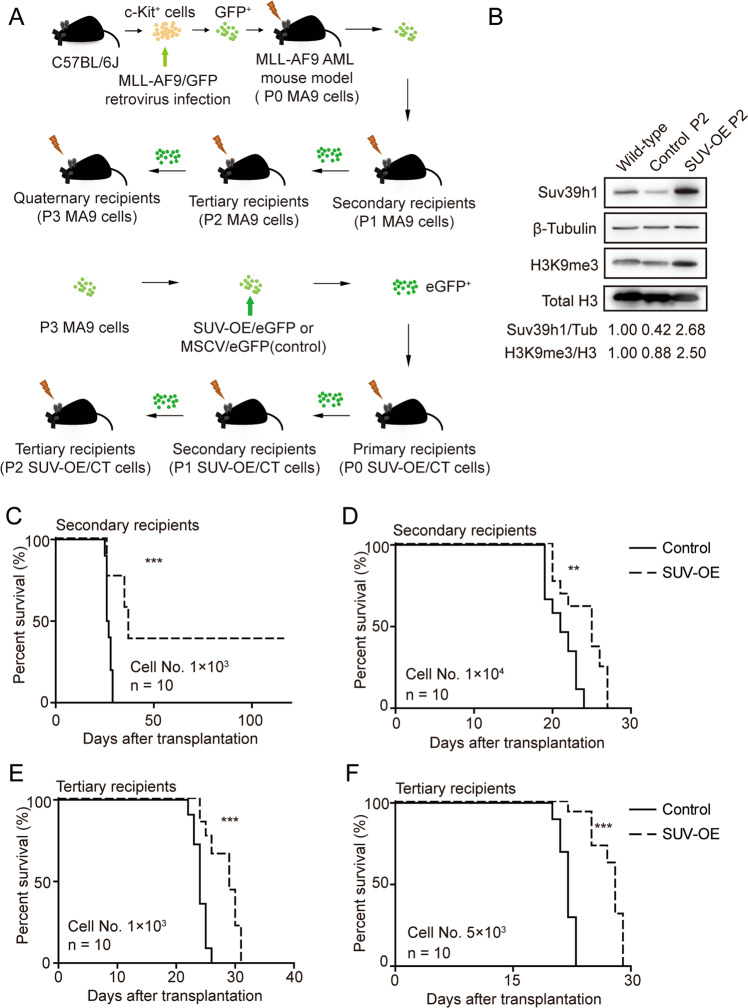
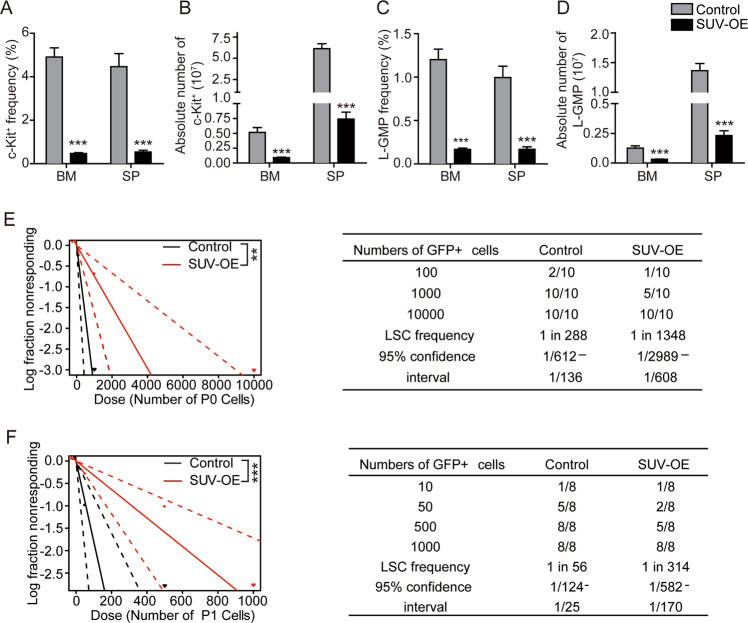
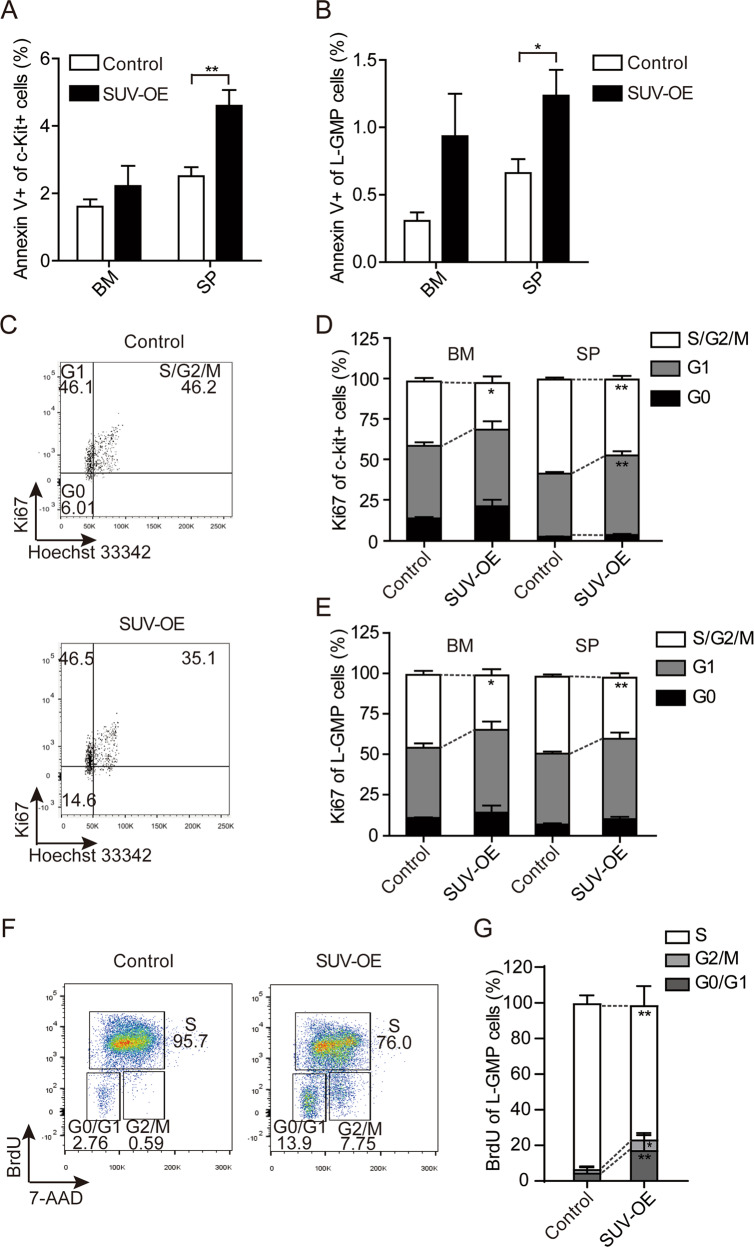
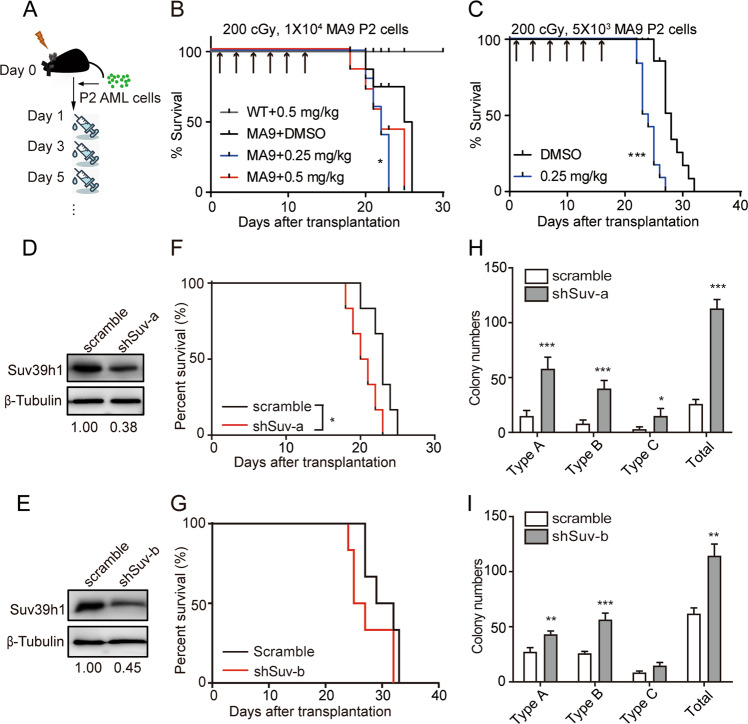
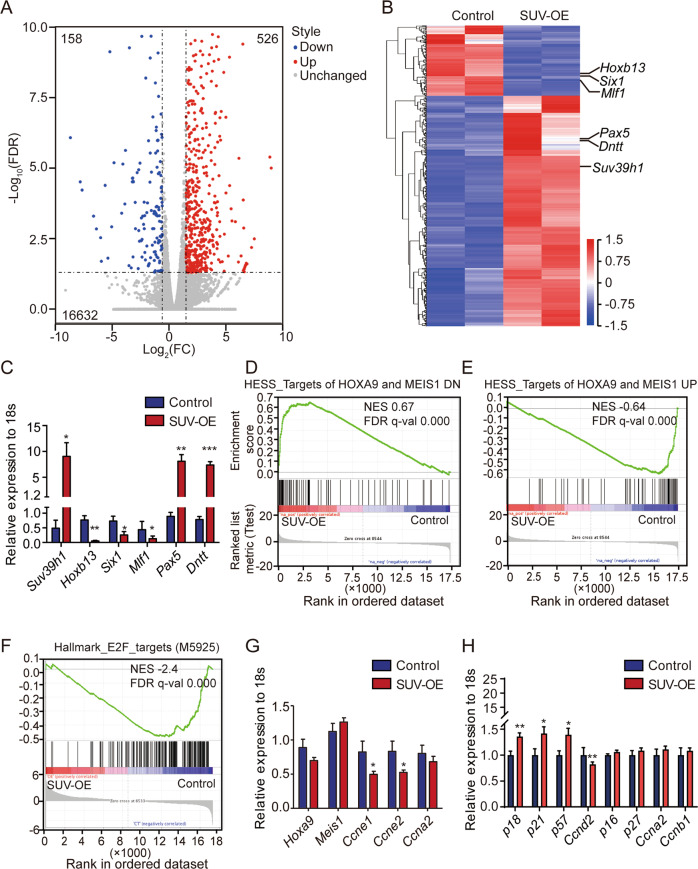
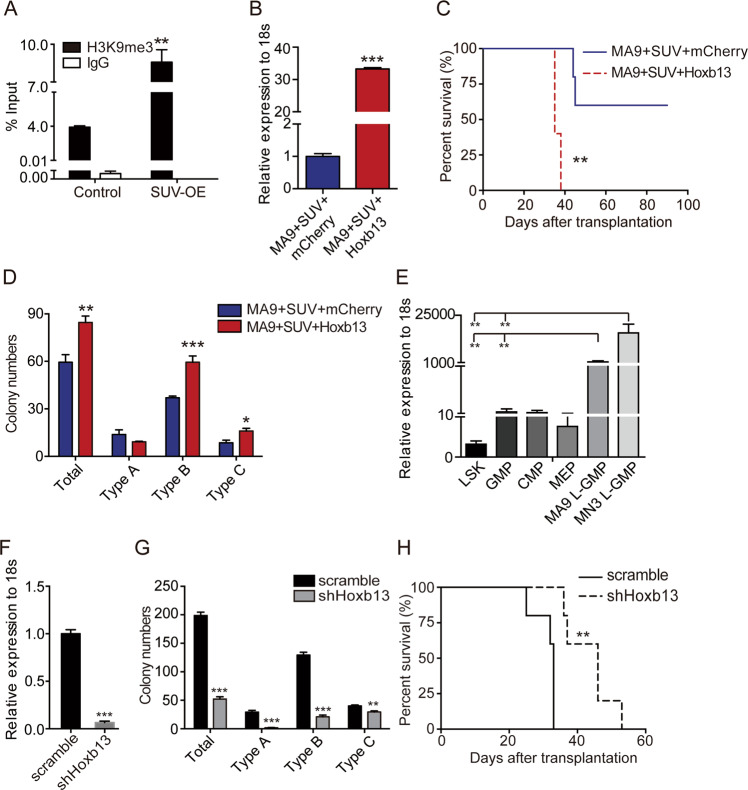
Epigenetic regulations play crucial roles in leukemogenesis and leukemia progression. SUV39H1 is the dominant H3K9 methyltransferase in the hematopoietic system, and its expression declines with aging. However, the role of SUV39H1 via its-mediated repressive modification H3K9me3 in leukemogenesis/leukemia progression remains to be explored. We found that SUV39H1 was down-regulated in a variety of leukemias, including MLL-r AML, as compared with normal individuals. Decreased levels of Suv39h1 expression and genomic H3K9me3 occupancy were observed in LSCs from MLL-r-induced AML mouse models in comparison with that of hematopoietic stem/progenitor cells. Suv39h1 overexpression increased leukemia latency and decreased the frequency of LSCs in MLL-r AML mouse models, while Suv39h1 knockdown accelerated disease progression with increased number of LSCs. Increased Suv39h1 expression led to the inactivation of Hoxb13 and Six1, as well as reversion of Hoxa9/Meis1 downstream target genes, which in turn decelerated leukemia progression. Interestingly, Hoxb13 expression is up-regulated in MLL-AF9-induced AML cells, while knockdown of Hoxb13 in MLL-AF9 leukemic cells significantly prolonged the survival of leukemic mice with reduced LSC frequencies. Our data revealed that SUV39H1 functions as a tumor suppressor in MLL-AF9-induced AML progression. These findings provide the direct link of SUV39H1 to AML development and progression.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
PBX3 is essential for leukemia stem cell maintenance in MLL-rearranged leukemia.Int J Cancer. 2017 Jul 15;141(2):324-335. doi: 10.1002/ijc.30739. Epub 2017 May 8. Int J Cancer. 2017. PMID: 28411381
-
SETDB1 mediated histone H3 lysine 9 methylation suppresses MLL-fusion target expression and leukemic transformation.Haematologica. 2020 Sep 1;105(9):2273-2285. doi: 10.3324/haematol.2019.223883. Haematologica. 2020. PMID: 33054052 Free PMC article.
-
ZNF521 Enhances MLL-AF9-Dependent Hematopoietic Stem Cell Transformation in Acute Myeloid Leukemias by Altering the Gene Expression Landscape.Int J Mol Sci. 2021 Oct 6;22(19):10814. doi: 10.3390/ijms221910814. Int J Mol Sci. 2021. PMID: 34639154 Free PMC article.
-
Learning from mouse models of MLL fusion gene-driven acute leukemia.Biochim Biophys Acta Gene Regul Mech. 2020 Aug;1863(8):194550. doi: 10.1016/j.bbagrm.2020.194550. Epub 2020 Apr 19. Biochim Biophys Acta Gene Regul Mech. 2020. PMID: 32320749 Review.
-
Exploring the contribution of Zfp521/ZNF521 on primary hematopoietic stem/progenitor cells and leukemia progression.Cell Tissue Res. 2024 Dec;398(3):161-173. doi: 10.1007/s00441-024-03926-2. Epub 2024 Oct 22. Cell Tissue Res. 2024. PMID: 39436449 Free PMC article. Review.
Cited by
-
Sox13 and M2-like leukemia-associated macrophages contribute to endogenous IL-34 caused accelerated progression of acute myeloid leukemia.Cell Death Dis. 2023 May 6;14(5):308. doi: 10.1038/s41419-023-05822-z. Cell Death Dis. 2023. PMID: 37149693 Free PMC article.
-
SUV39H1 epigenetically modulates the MCPIP1-AURKA signaling axis to enhance neuroblastoma tumorigenesis.Oncogene. 2024 Nov;43(45):3306-3320. doi: 10.1038/s41388-024-03164-4. Epub 2024 Sep 19. Oncogene. 2024. PMID: 39300256 Free PMC article.
-
SUV39H1 is a novel biomarker targeting oxidative phosphorylation in hepatitis B virus-associated hepatocellular carcinoma.BMC Cancer. 2023 Nov 28;23(1):1159. doi: 10.1186/s12885-023-11633-4. BMC Cancer. 2023. PMID: 38017386 Free PMC article.
-
Circulating ESR1, long non-coding RNA HOTAIR and microRNA-130a gene expression as biomarkers for breast cancer stage and metastasis.Sci Rep. 2023 Dec 19;13(1):22654. doi: 10.1038/s41598-023-50007-5. Sci Rep. 2023. PMID: 38114755 Free PMC article.
-
HOXB13 in cancer development: molecular mechanisms and clinical implications.Front Med. 2025 Jun;19(3):439-455. doi: 10.1007/s11684-024-1119-x. Epub 2025 Mar 11. Front Med. 2025. PMID: 40067581 Review.
References
-
- Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW. SnapShot: acute myeloid leukemia. Cancer Cell. 2012;22:698–698. - PubMed
-
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
