

Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue
- PMID: 33038072
- PMCID: PMC7547588
- DOI: 10.14814/phy2.14607
Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue
Abstract
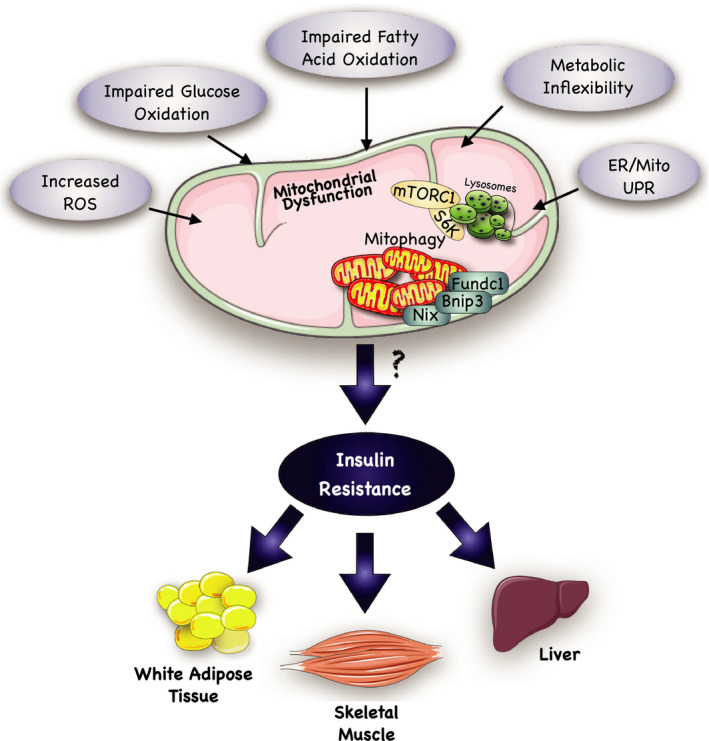
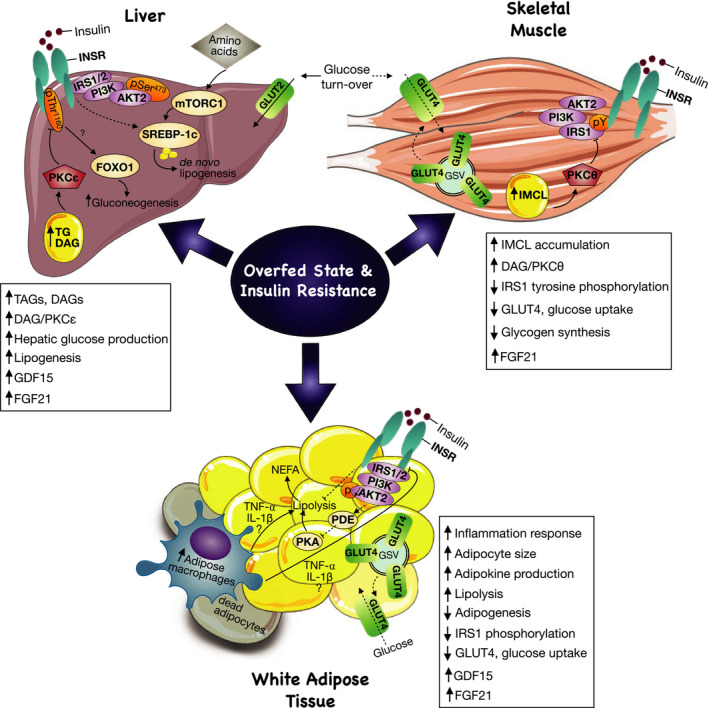
Insulin resistance is a metabolic disorder affecting multiple tissues and is a precursor event to type 2 diabetes (T2D). As T2D affects over 425 million people globally, there is an imperative need for research into insulin resistance to better understand the underlying mechanisms. The proposed mechanisms involved in insulin resistance include both whole body aspects, such as inflammation and metabolic inflexibility; as well as cellular phenomena, such as lipotoxicity, ER stress, and mitochondrial dysfunction. Despite numerous studies emphasizing the role of lipotoxicity in the pathogenesis of insulin resistance, an understanding of the interplay between tissues and these proposed mechanisms is still emerging. Furthermore, the tissue-specific and unique responses each of the three major insulin target tissues and how each interconnect to regulate the whole body insulin response has become a new priority in metabolic research. With an emphasis on skeletal muscle, this mini-review highlights key similarities and differences in insulin signaling and resistance between different target-tissues, and presents the latest findings related to how these tissues communicate to control whole body metabolism.
Keywords: adipose tissue; insulin resistance; lipotoxicity; liver; mitochondrial dysfunction; skeletal muscle.
© 2020 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of The Physiological Society and the American Physiological Society.
Conflict of interest statement
The authors do not have any conflicts of interest to declare.
Figures
References
-
- Aguirre, V. , Uchida, T. , Yenush, L. , Davis, R. , & White, M. F. (2000). The c‐Jun NH2‐terminal kinase promotes insulin resistance during association with insulin receptor substrate‐1 and phosphorylation of Ser307. Journal of Biological Chemistry, 275, 9047–9054. 10.1074/jbc.275.12.9047 - DOI - PubMed
-
- Albers, P. H. , Pedersen, A. J. T. , Birk, J. B. , Kristensen, D. E. , Vind, B. F. , Baba, O. , Nøhr, J. , Højlund, K. , & Wojtaszewski, J. F. P. (2015). Human muscle fiber type‐specific insulin signaling: impact of obesity and type 2 diabetes. Diabetes, 64, 485–497. 10.2337/db14-0590 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
