

Pan-cancer analysis of somatic mutations in miRNA genes
- PMID: 33038763
- PMCID: PMC7648123
- DOI: 10.1016/j.ebiom.2020.103051
Pan-cancer analysis of somatic mutations in miRNA genes
Abstract
Background: miRNAs are considered important players in oncogenesis, serving either as oncomiRs or suppressormiRs. Although the accumulation of somatic alterations is an intrinsic aspect of cancer development and many important cancer-driving mutations have been identified in protein-coding genes, the area of functional somatic mutations in miRNA genes is heavily understudied.
Methods: Here, based on the analysis of large genomic datasets, mostly the whole-exome sequencing of over 10,000 cancer/normal sample pairs deposited within the TCGA repository, we undertook an analysis of somatic mutations in miRNA genes.
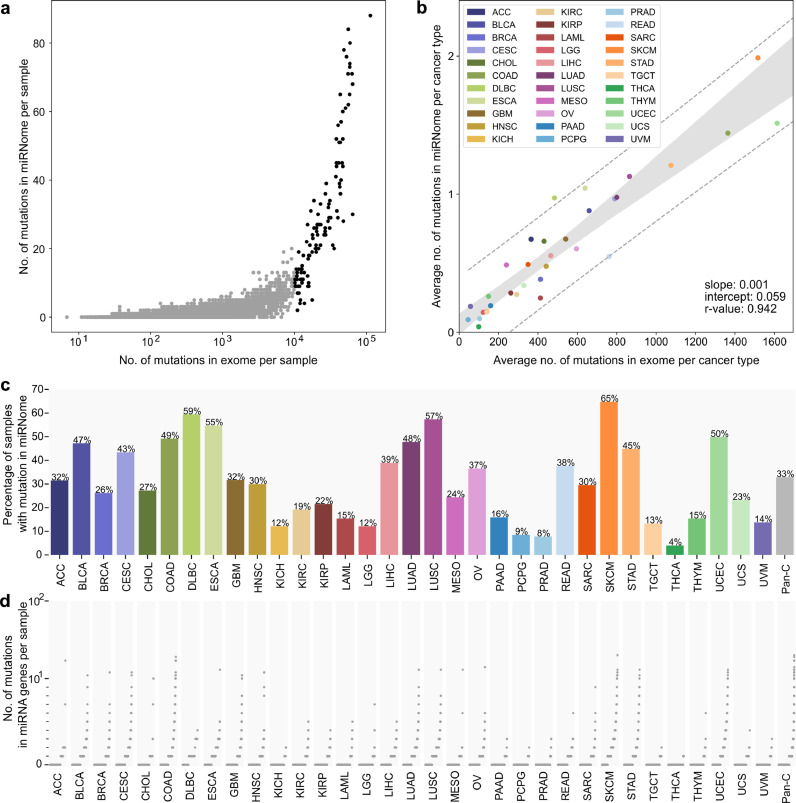
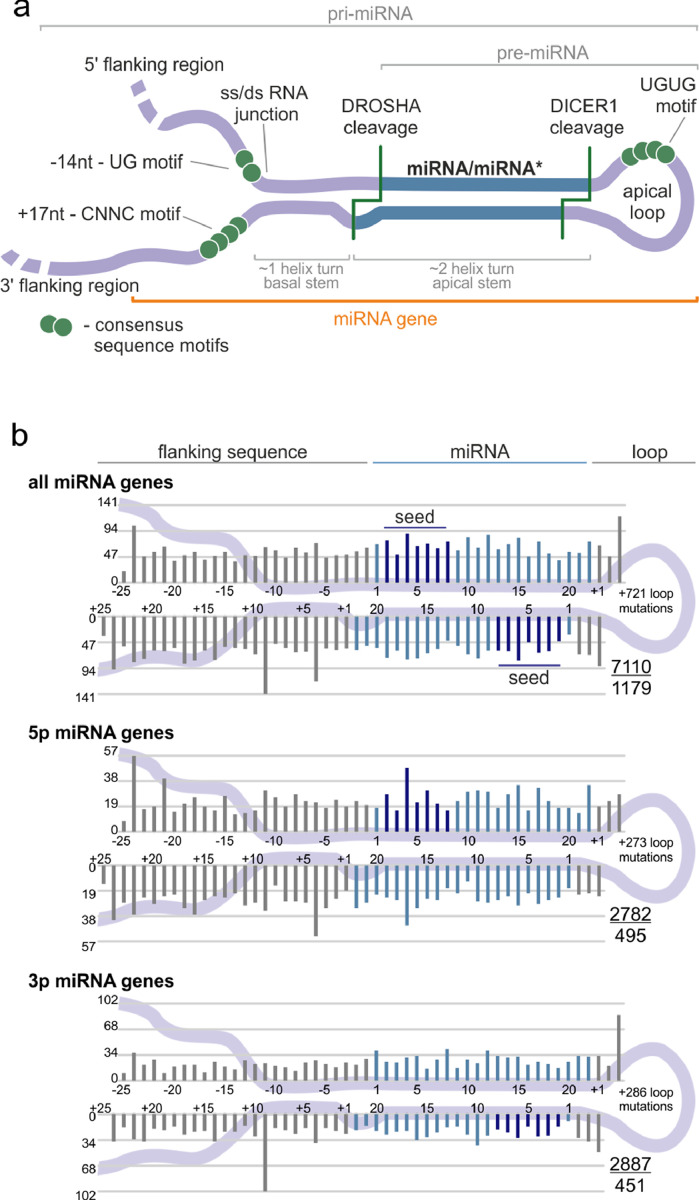
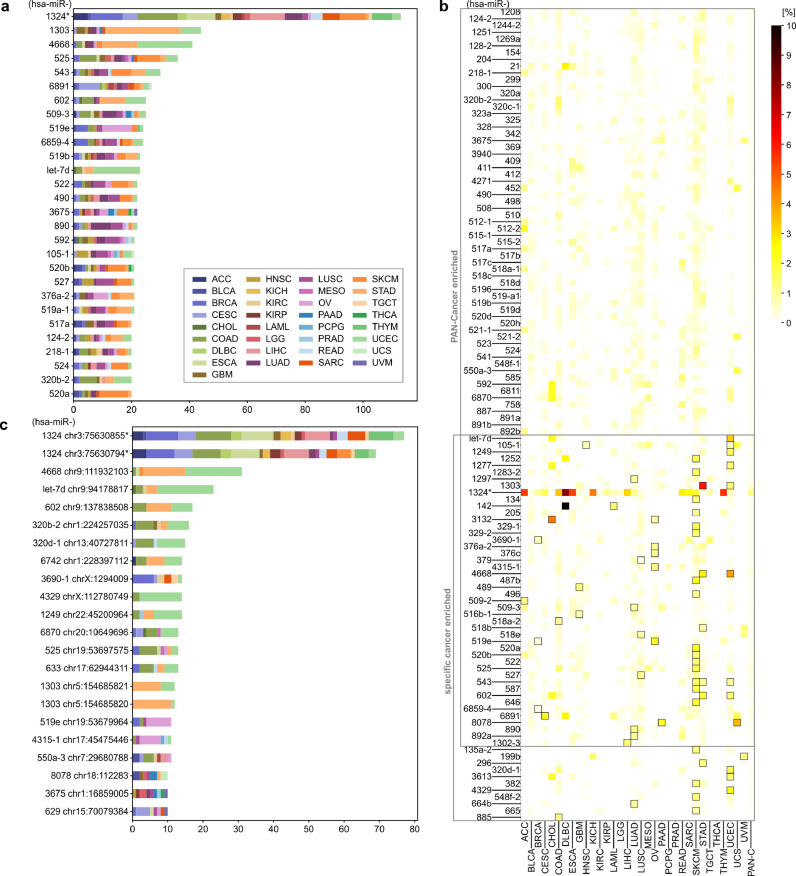
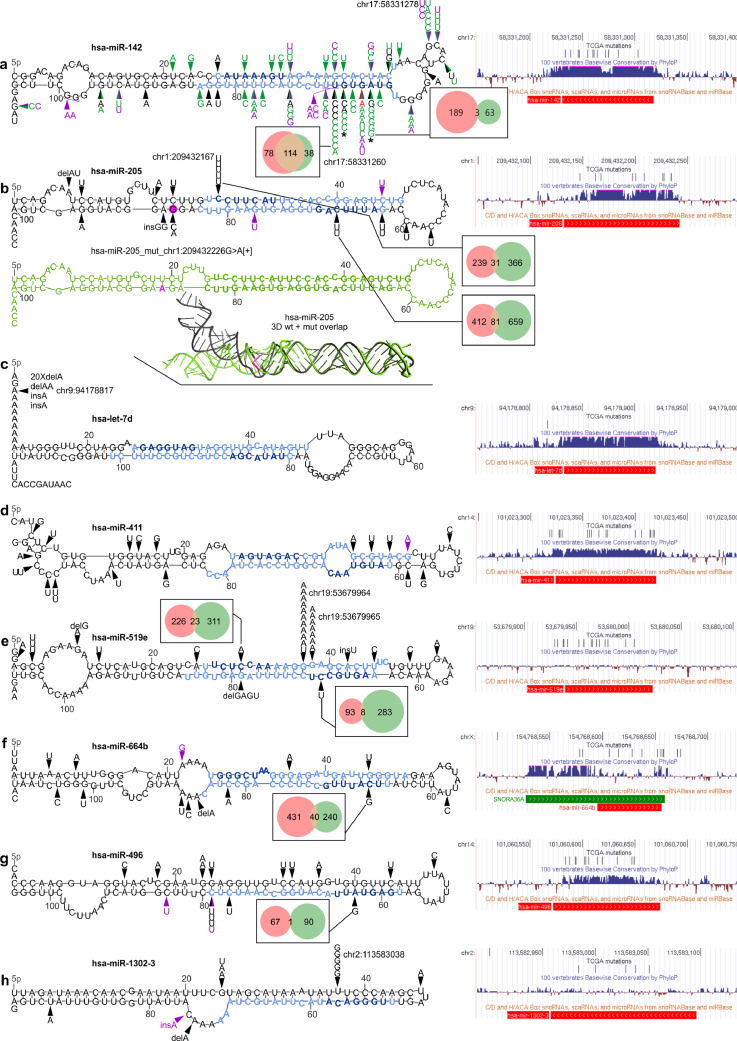
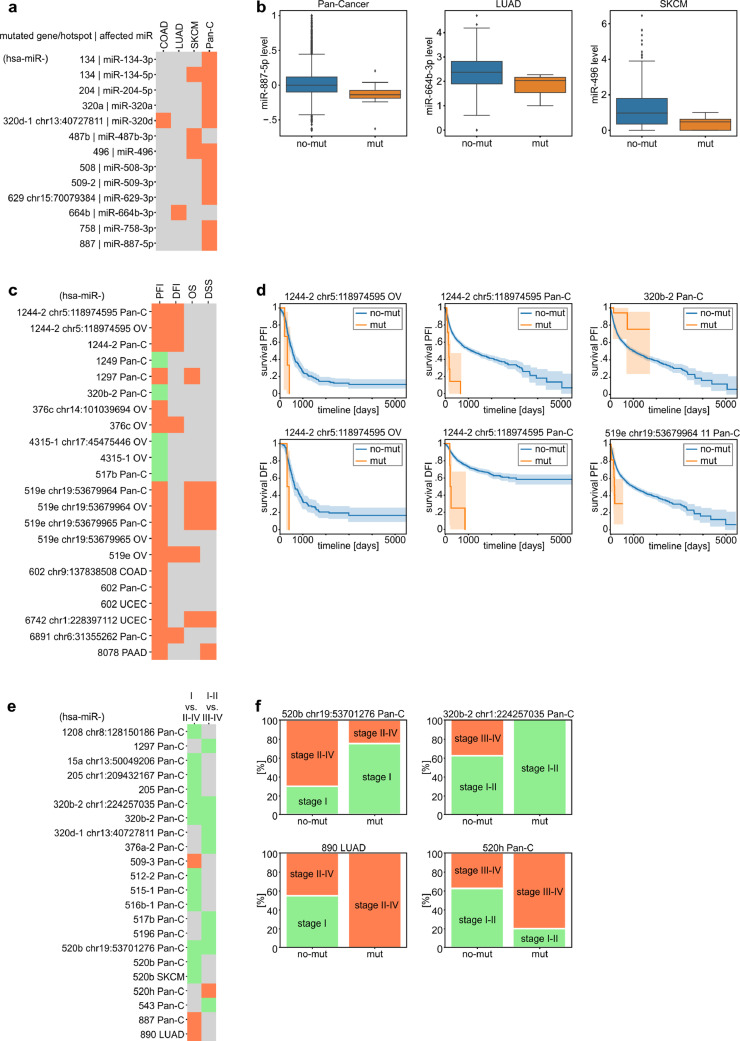
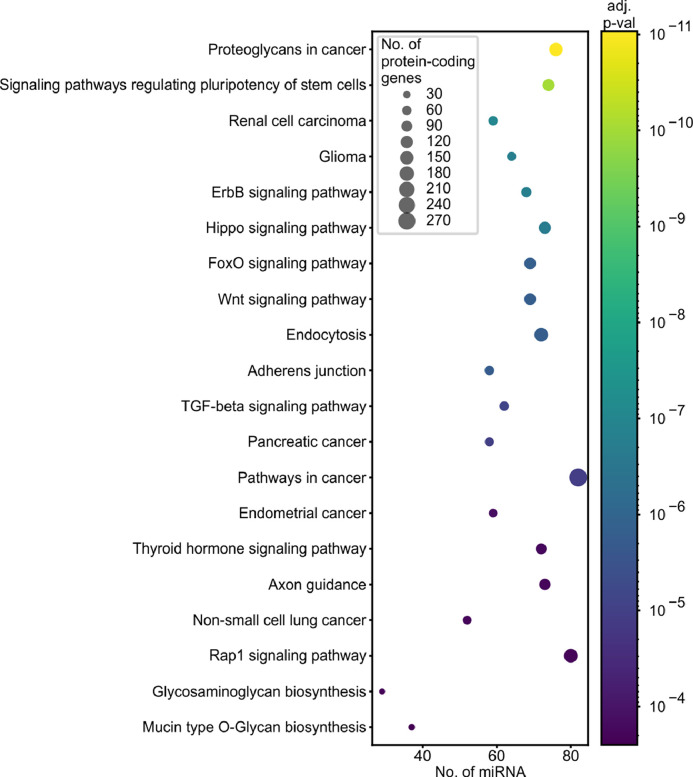
Findings: We identified and characterized over 10,000 somatic mutations and showed that some of the miRNA genes are overmutated in Pan-Cancer and/or specific cancers. Nonrandom occurrence of the identified mutations was confirmed by a strong association of overmutated miRNA genes with KEGG pathways, most of which were related to specific cancer types or cancer-related processes. Additionally, we showed that mutations in some of the overmutated genes correlate with miRNA expression, cancer staging, and patient survival.
Interpretation: Our study is the first comprehensive Pan-Cancer study of cancer somatic mutations in miRNA genes. It may help to understand the consequences of mutations in miRNA genes and the identification of miRNA functional mutations. The results may also be the first step (form the basis and provide the resources) in the development of computational and/or statistical approaches/tools dedicated to the identification of cancer-driver miRNA genes.
Funding: This work was supported by research grants from the Polish National Science Centre 2016/22/A/NZ2/00184 and 2015/17/N/NZ3/03629.
Keywords: Non-coding; Pan-Cancer; Somatic mutations; TCGA; miRNA.
Copyright © 2020 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare no competing financial interests.
Figures
References
-
- Horn S, Figl A, Rachakonda PS. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. - PubMed
-
- Vinagre J, Almeida A, Pópulo H. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. - PubMed
-
- Hosseinahli N, Aghapour M, Duijf PHG, Baradaran B. Treating cancer with microRNA replacement therapy: a literature review. J Cell Physiol. 2018;233:5574–5588. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
