

Efficient dynamic variation graphs
- PMID: 33040146
- PMCID: PMC7850124
- DOI: 10.1093/bioinformatics/btaa640
Efficient dynamic variation graphs
Abstract
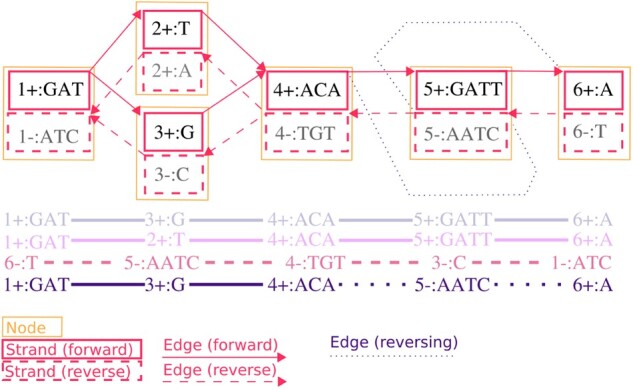
Motivation: Pangenomics is a growing field within computational genomics. Many pangenomic analyses use bidirected sequence graphs as their core data model. However, implementing and correctly using this data model can be difficult, and the scale of pangenomic datasets can be challenging to work at. These challenges have impeded progress in this field.
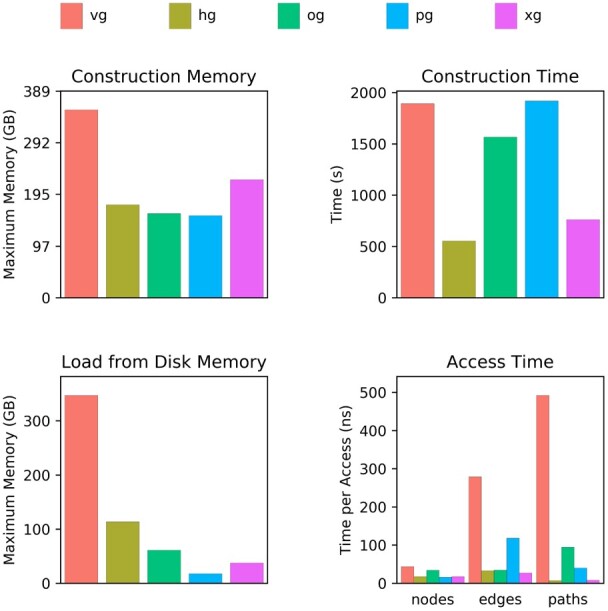
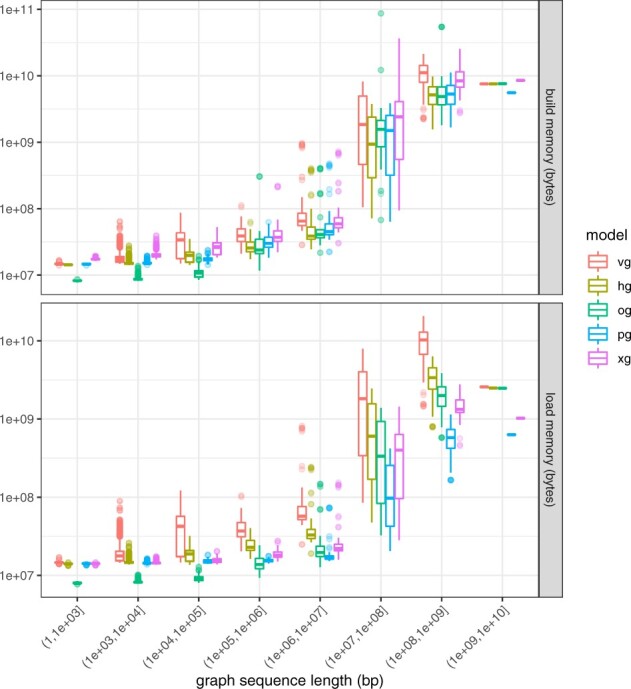
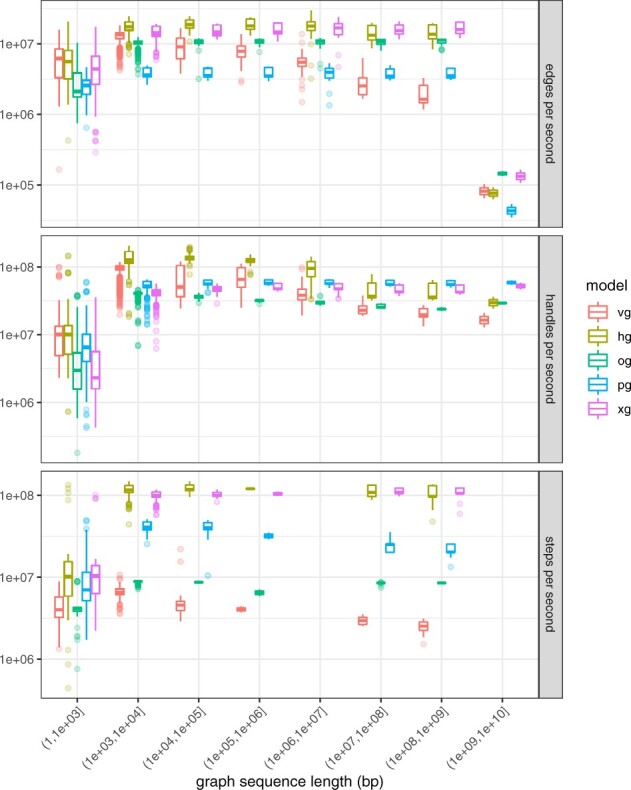
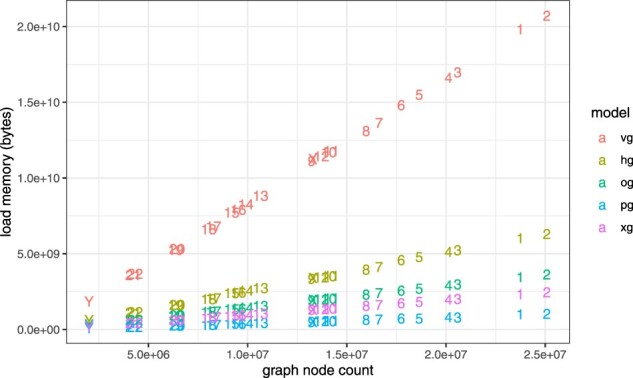
Results: Here, we present a stack of two C++ libraries, libbdsg and libhandlegraph, which use a simple, field-proven interface, designed to expose elementary features of these graphs while preventing common graph manipulation mistakes. The libraries also provide a Python binding. Using a diverse collection of pangenome graphs, we demonstrate that these tools allow for efficient construction and manipulation of large genome graphs with dense variation. For instance, the speed and memory usage are up to an order of magnitude better than the prior graph implementation in the VG toolkit, which has now transitioned to using libbdsg's implementations.
Availability and implementation: libhandlegraph and libbdsg are available under an MIT License from https://github.com/vgteam/libhandlegraph and https://github.com/vgteam/libbdsg.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Brehm W. (2019) Hash tables with pseudorandom global order. INFOCOMP J. Comput. Sci., 18, 20–25.
