

From Iron Chelation to Overload as a Therapeutic Strategy to Induce Ferroptosis in Leukemic Cells
- PMID: 33042852
- PMCID: PMC7530268
- DOI: 10.3389/fonc.2020.586530
From Iron Chelation to Overload as a Therapeutic Strategy to Induce Ferroptosis in Leukemic Cells
Abstract
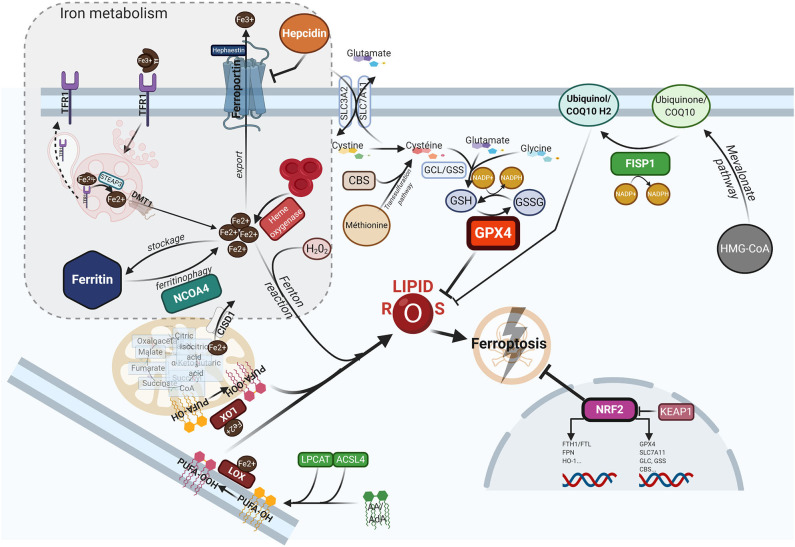
Despite its crucial importance in numerous physiological processes, iron also causes oxidative stress and damage which can promote the growth and proliferation of leukemic cells. Iron metabolism is strictly regulated and the related therapeutic approaches to date have been to restrict iron availability to tumor cells. However, since a new form of iron-catalyzed cell death has been described, termed ferroptosis, and subsequently better understood, iron excess is thought to represent an opportunity to selectively kill leukemic cells and spare normal hematopoietic cells, based on their differential iron needs. This review summarizes the physiology of iron metabolism and its deregulation in leukemia, the known ferrotoposis pathways, and therapeutic strategies to target the altered iron metabolism in leukemia for the purposes of initiating ferroptosis in these cancer cells.
Keywords: acute myeloid leukemia; ferritinophagy; ferroptosis; iron; reactive oxygen species.
Copyright © 2020 Grignano, Birsen, Chapuis and Bouscary.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources