

Alzheimer's Disease Prevention and Treatment: Case for Optimism
- PMID: 33043322
- PMCID: PMC7546530
- DOI: 10.33597/aimm.02-1008
Alzheimer's Disease Prevention and Treatment: Case for Optimism
Abstract
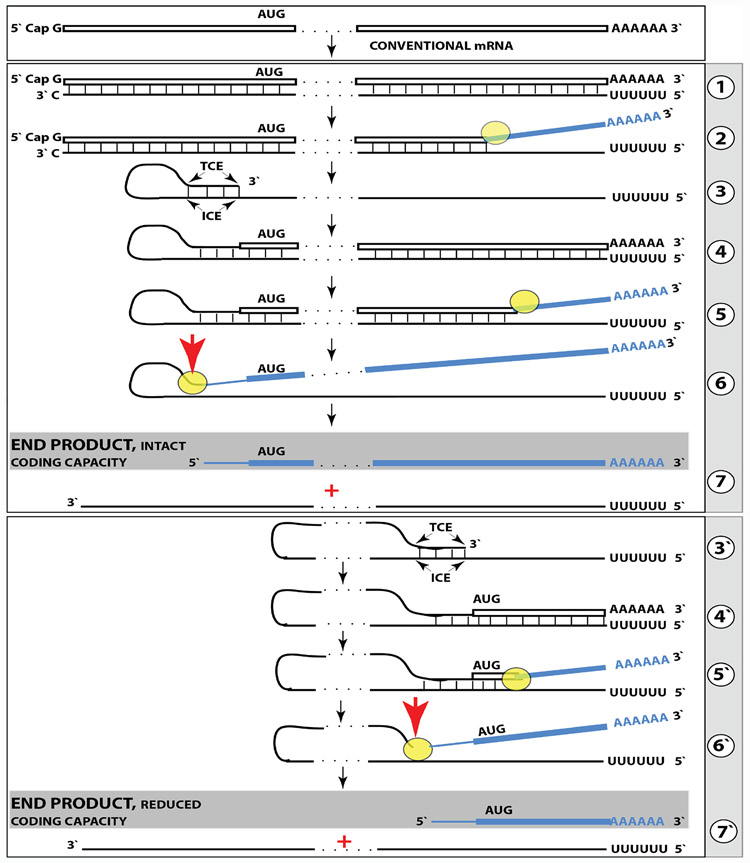
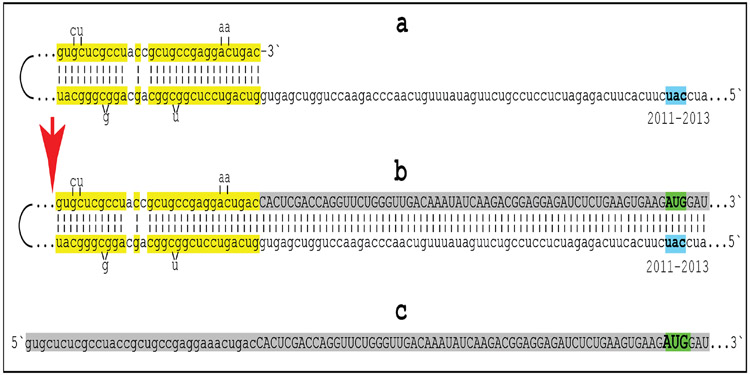
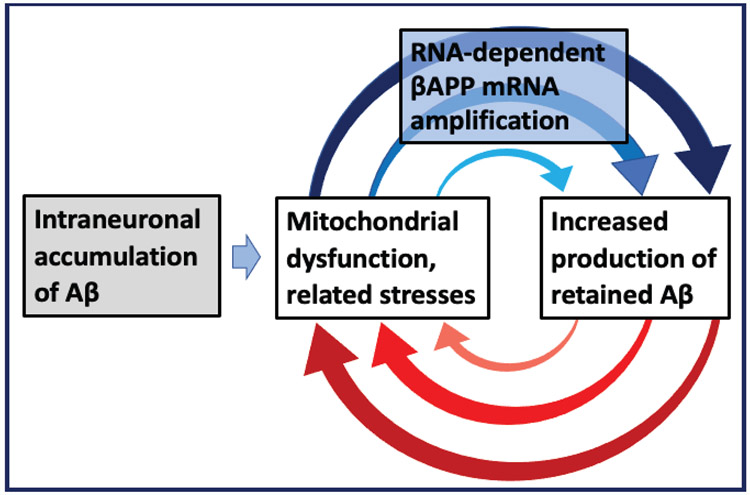
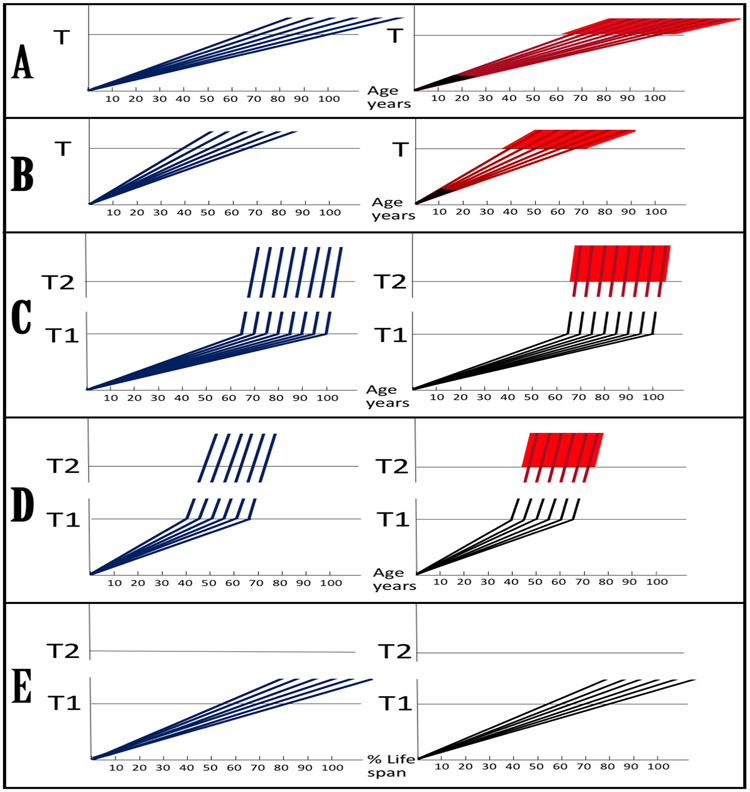
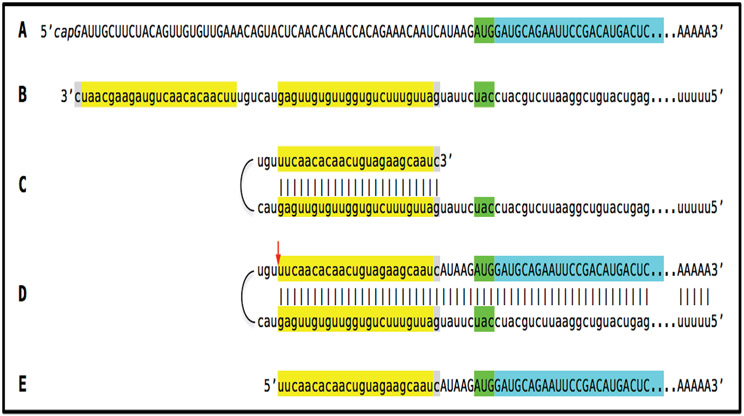
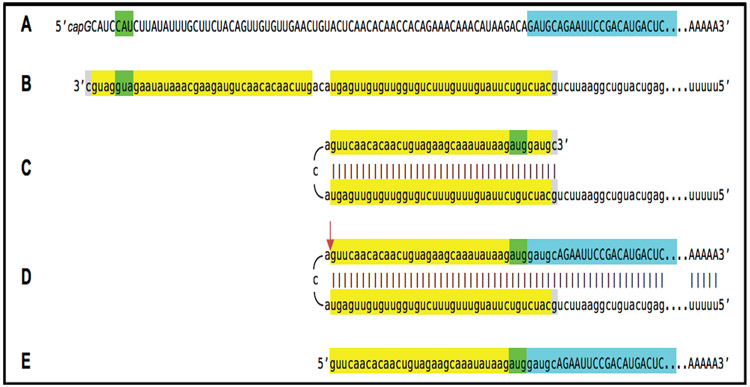
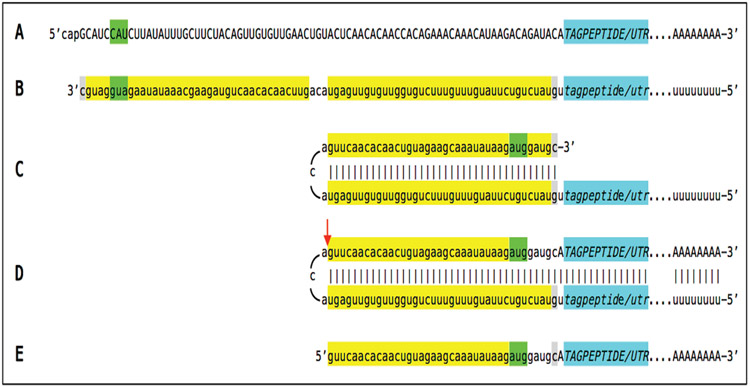
A paradigm shift is under way in the Alzheimer's field. A view of Alzheimer's disease, AD, prevailing until now, the old paradigm, maintains that it is initiated and driven by the overproduction and extracellular accumulation of beta-amyloid, Aβ; a peptide assumed to be derived, both in health and disease, solely by proteolysis of its large precursor, βAPP. In AD, according to this view, Aβ overproduction-associated neurodegeneration begins early, accumulates throughout the lifespan, and manifests symptomatically late in life. A number of drugs, designed within the framework of exceptionality of the βAPP proteolytic/secretory pathway in Aβ production in Alzheimer's disease, achieved spectacular successes in treatment, even the reversal, of AD symptoms in animal models. Without exception, they all exhibited equally spectacular failures in human clinical trials. This paradigm provides few causes for optimism with regard to prevention and treatment of AD. In its context, the disease is considered untreatable in the symptomatic phase; even prodromal cases are assumed too advanced for treatment because Aβ-triggered damages have been accumulating for preceding decades, presumably starting in the early twenties and, to be effective, this is when therapeutic intervention should commence and continue for life. The new paradigm does not dispute the seminal role of Aβ in AD but posits that beta-amyloid produced in the βAPP proteolytic/secretory pathway causes AD in humans no more than it does in non-human mammals that share this pathway with humans, accumulate Aβ as they age, but do not develop the disease. Alzheimer's disease, according to this outlook, is driven by the AD-specific pathway of Aβ production, independent of βAPP and absent in animals. Its activation, late in life, occurs through accumulation, via both cellular uptake of secreted Aβ and neuronal retention of a fraction of beta-amyloid produced in the βAPP proteolytic pathway, of intraneuronal Aβ, which triggers mitochondrial dysfunction. Cellular stresses associated with mitochondrial dysfunction, or, probably, the integrated stress response, ISR, elicited by it, activate an AD-specific Aβ production pathway. In it, every conventionally produced βAPP mRNA molecule potentially serves repeatedly as a template for production of severely 5'-truncated mRNA encoding C99 fragment of βAPP, the immediate precursor of Aβ that is processed in a non-secretory pathway, apparently in a neuron-specific manner. The resulting intraneuronally retained Aβ augments mitochondrial dysfunction, which, in turn, sustains the activity of the βAPP mRNA amplification pathway. These self-propagating Aβ overproduction/mitochondrial dysfunction mutual feedback cycles constitute the engine that drives AD and ultimately triggers neuronal death. In this paradigm, preventive treatment can be initiated any time prior to commencement of βAPP mRNA amplification. Moreover, there are good reasons to believe that with a drug blocking the amplification pathway, it would be possible not only to preempt the disease but also stop and reverse it even when early AD symptoms are already manifested. Thus, the new paradigm introduces a novel theory of Alzheimer's disease. It explains the observed discordances, determines defined therapeutic targets, provides blueprints for a new generation of conceptually distinct AD models and specifies design of a reporter for the mRNA amplification pathway. Most importantly, it offers detailed guidance and tangible hope for prevention of the disease and its treatment at the early symptomatic stages.
Keywords: AD models for the new paradigm; Asymmetric RNA-dependent βAPP mRNA amplification; Intraneuronal retention of Aβ; Reporter-based optimal AD models; Universal reporter for the mammalian RNA-dependent mRNA amplification process; βAPP-independent generation of Aβ.
Conflict of interest statement
Conflicts of Interest Authors declare no conflict of interest.
Figures
References
-
- Conan-Doyle A. The sign of the four. Lippincott’s Monthly Magazine. 1890.
-
- Volloch V. A mechanism for ß-amyloid overproduction in Alzheimer's disease: Precursor-independent generation of ß-amyloid via antisense RNA-primed mRNA synthesis. FEBS Lett. 1996;390:124–8. - PubMed
-
- Volloch V. Mechanism for ß-amyloid overproduction in sporadic Alzheimer’s Disease: Possible antisense RNA-mediated generation of a 5’-truncated ßAPP mRNA encoding 12 kDa C-terminal fragment of ßAPP, the immediate precursor of Aß In: Molecular Mechanisms of Dementia. 1997, Wasco W and Tanzi R, Eds.
-
- Volloch V. Possible mechanism for resistance to Alzheimer's disease (AD) in mice suggests new approach to generate a mouse model for sporadic AD and may explain familial resistance to AD in man. Exp Neurobiol. 1997;144:214–8. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources