

BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation
- PMID: 33044904
- PMCID: PMC8496715
- DOI: 10.1080/15548627.2020.1821548
BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation
Abstract
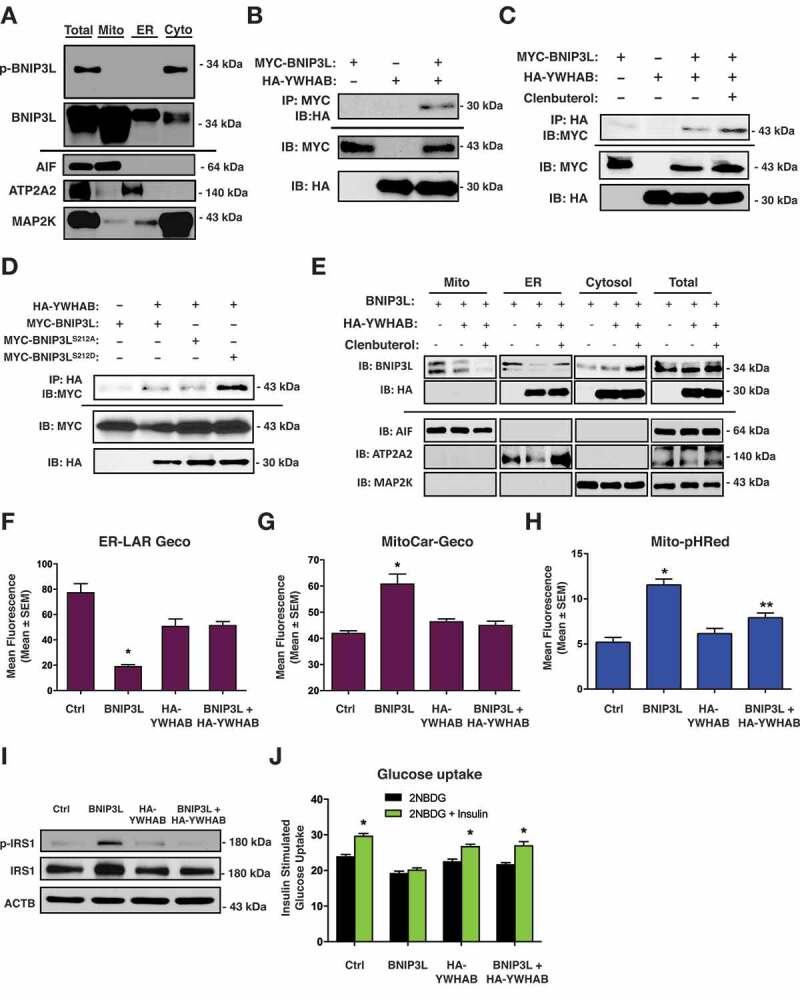
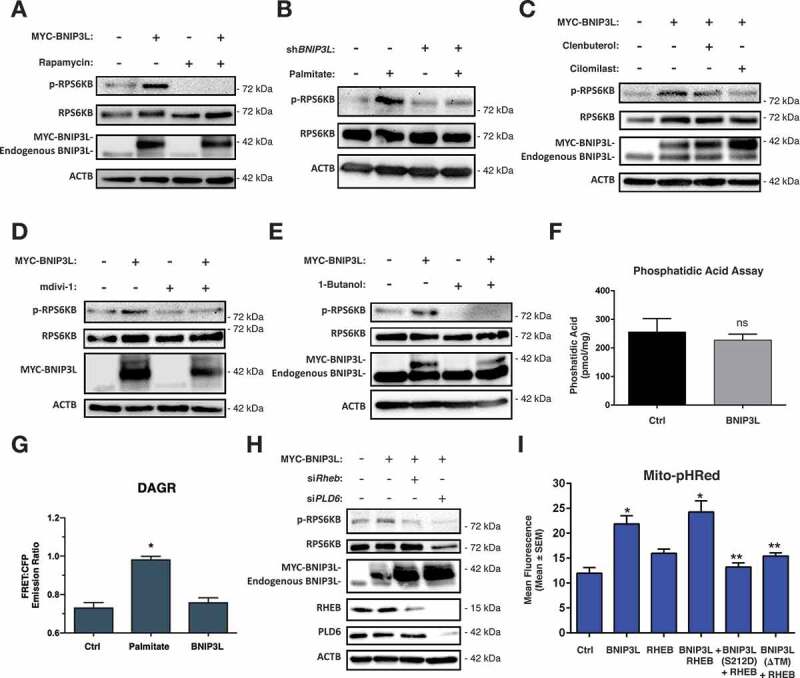
Lipotoxicity is a form of cellular stress caused by the accumulation of lipids resulting in mitochondrial dysfunction and insulin resistance in muscle. Previously, we demonstrated that the mitophagy receptor BNIP3L/Nix is responsive to lipotoxicity and accumulates in response to a high-fat (HF) feeding. To provide a better understanding of this observation, we undertook gene expression array and shot-gun metabolomics studies in soleus muscle from rodents on an HF diet. Interestingly, we observed a modest reduction in several autophagy-related genes. Moreover, we observed alterations in the fatty acyl composition of cardiolipins and phosphatidic acids. Given the reported roles of these phospholipids and BNIP3L in mitochondrial dynamics, we investigated aberrant mitochondrial turnover as a mechanism of impaired myocyte insulin signaling. In a series of gain-of-function and loss-of-function experiments in rodent and human myotubes, we demonstrate that BNIP3L accumulation triggers mitochondrial depolarization, calcium-dependent activation of DNM1L/DRP1, and mitophagy. In addition, BNIP3L can inhibit insulin signaling through activation of MTOR-RPS6KB/p70S6 kinase inhibition of IRS1, which is contingent on phosphatidic acids and RHEB. Finally, we demonstrate that BNIP3L-induced mitophagy and impaired glucose uptake can be reversed by direct phosphorylation of BNIP3L by PRKA/PKA, leading to the translocation of BNIP3L from the mitochondria and sarcoplasmic reticulum to the cytosol. These findings provide insight into the role of BNIP3L, mitochondrial turnover, and impaired myocyte insulin signaling during an overfed state when overall autophagy-related gene expression is reduced. Furthermore, our data suggest a mechanism by which exercise or pharmacological activation of PRKA may overcome myocyte insulin resistance.Abbreviations: BCL2: B cell leukemia/lymphoma 2; BNIP3L/Nix: BCL2/adenovirus E1B interacting protein 3-like; DNM1L/DRP1: dynamin 1-like; FUNDC1: FUN14 domain containing 1; IRS1: insulin receptor substrate 1; MAP1LC3A/LC3: microtubule-associated protein 1 light chain 3 alpha; MFN1: mitofusin 1; MFN2: mitofusin 2; MTOR: mechanistic target of rapamycin kinase; OPA1: OPA1 mitochondrial dynamin like GTPase; PDE4i: phosphodiesterase 4 inhibitor; PLD1: phospholipase D1; PLD6: phospholipase D family member 6; PRKA/PKA: protein kinase, AMP-activated; PRKCD/PKCδ: protein kinase C, delta; PRKCQ/PKCθ: protein kinase C, theta; RHEB: Ras homolog enriched in brain; RPS6KB/p70S6K: ribosomal protein S6 kinase; SQSTM1/p62: sequestosome 1; YWHAB/14-3-3β: tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein beta.
Keywords: Insulin signaling; MTOR; Nix; PKA; mitochondria; mitophagy; muscle.
Conflict of interest statement
No potential conflict of interest was reported by the authors.
Figures
Similar articles
-
BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation.Autophagy. 2019 Jul;15(7):1182-1198. doi: 10.1080/15548627.2019.1580095. Epub 2019 Feb 22. Autophagy. 2019. PMID: 30741592 Free PMC article.
-
BNIP3L-mediated mitophagy is required for mitochondrial remodeling during the differentiation of optic nerve oligodendrocytes.Autophagy. 2021 Oct;17(10):3140-3159. doi: 10.1080/15548627.2020.1871204. Epub 2021 Jan 19. Autophagy. 2021. PMID: 33404293 Free PMC article.
-
Emerging views of mitophagy in immunity and autoimmune diseases.Autophagy. 2020 Jan;16(1):3-17. doi: 10.1080/15548627.2019.1603547. Epub 2019 Apr 21. Autophagy. 2020. PMID: 30951392 Free PMC article.
-
Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles.Autophagy. 2021 Feb;17(2):385-401. doi: 10.1080/15548627.2020.1725377. Epub 2020 Feb 12. Autophagy. 2021. PMID: 32048886 Free PMC article. Review.
-
A brief overview of BNIP3L/NIX receptor-mediated mitophagy.FEBS Open Bio. 2021 Dec;11(12):3230-3236. doi: 10.1002/2211-5463.13307. Epub 2021 Oct 11. FEBS Open Bio. 2021. PMID: 34597467 Free PMC article. Review.
Cited by
-
Ablation of Shank3 alleviates cardiac dysfunction in aging mice by promoting CaMKII activation and Parkin-mediated mitophagy.Redox Biol. 2022 Dec;58:102537. doi: 10.1016/j.redox.2022.102537. Epub 2022 Nov 18. Redox Biol. 2022. PMID: 36436456 Free PMC article.
-
PINK1 and Parkin regulate IP3R-mediated ER calcium release.Nat Commun. 2023 Aug 25;14(1):5202. doi: 10.1038/s41467-023-40929-z. Nat Commun. 2023. PMID: 37626046 Free PMC article.
-
Mitophagy: A potential therapeutic target for insulin resistance.Front Physiol. 2022 Aug 23;13:957968. doi: 10.3389/fphys.2022.957968. eCollection 2022. Front Physiol. 2022. PMID: 36082218 Free PMC article. Review.
-
Mitophagy in metabolic syndrome.J Clin Hypertens (Greenwich). 2023 May;25(5):397-403. doi: 10.1111/jch.14650. Epub 2023 Apr 11. J Clin Hypertens (Greenwich). 2023. PMID: 37039609 Free PMC article. Review.
-
Multiple-Tissue and Multilevel Analysis on Differentially Expressed Genes and Differentially Correlated Gene Pairs for HFpEF.Front Genet. 2021 Jul 8;12:668702. doi: 10.3389/fgene.2021.668702. eCollection 2021. Front Genet. 2021. PMID: 34306013 Free PMC article.
References
-
- Gordon JW, Dolinsky VW, Mughal W, et al. Targeting skeletal muscle mitochondria to prevent type 2 diabetes in youth. Biochem Cell Biol. 2015;93:452–465. - PubMed
-
- Hesselink MKC, Schrauwen-Hinderling V, Schrauwen P.. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol. 2016;12:633–645. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous