

ΦX174 Attenuation by Whole-Genome Codon Deoptimization
- PMID: 33045052
- PMCID: PMC7881332
- DOI: 10.1093/gbe/evaa214
ΦX174 Attenuation by Whole-Genome Codon Deoptimization
Abstract
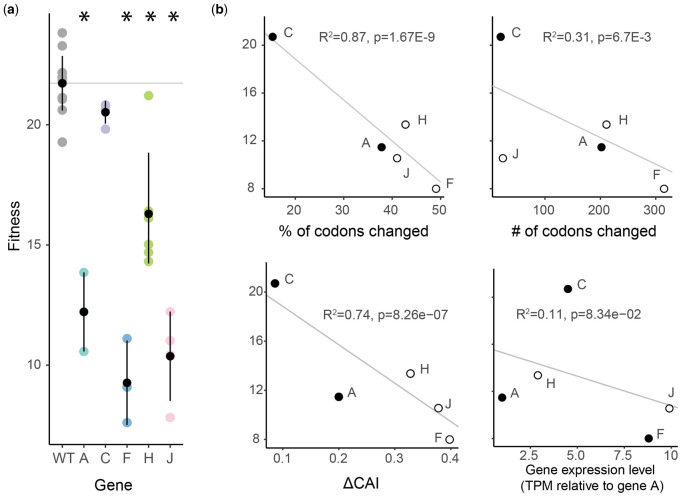
Natural selection acting on synonymous mutations in protein-coding genes influences genome composition and evolution. In viruses, introducing synonymous mutations in genes encoding structural proteins can drastically reduce viral growth, providing a means to generate potent, live-attenuated vaccine candidates. However, an improved understanding of what compositional features are under selection and how combinations of synonymous mutations affect viral growth is needed to predictably attenuate viruses and make them resistant to reversion. We systematically recoded all nonoverlapping genes of the bacteriophage ΦX174 with codons rarely used in its Escherichia coli host. The fitness of recombinant viruses decreases as additional deoptimizing mutations are made to the genome, although not always linearly, and not consistently across genes. Combining deoptimizing mutations may reduce viral fitness more or less than expected from the effect size of the constituent mutations and we point out difficulties in untangling correlated compositional features. We test our model by optimizing the same genes and find that the relationship between codon usage and fitness does not hold for optimization, suggesting that wild-type ΦX174 is at a fitness optimum. This work highlights the need to better understand how selection acts on patterns of synonymous codon usage across the genome and provides a convenient system to investigate the genetic determinants of virulence.
Keywords: bacteriophage; codon bias; epistasis; fitness landscape; live-attenuated vaccine; synthetic biology.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures





References
-
- Bailey SF, Hinz A, Kassen R. 2014. Adaptive synonymous mutations in an experimentally evolved Pseudomonas fluorescens population. Nat Commun. 5:4076. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
