

Type 1 tyrosinemia in Finland: a nationwide study
- PMID: 33046095
- PMCID: PMC7549233
- DOI: 10.1186/s13023-020-01547-w
Type 1 tyrosinemia in Finland: a nationwide study
Abstract
Background: Introduction of nitisinone and newborn screening (NBS) have transformed the treatment of type 1 tyrosinemia, but the effects of these changes on the long-term outcomes remain obscure. Also, the predictors for later complications, the significance of drug levels and the normalization of laboratory and imaging findings are poorly known. We investigated these issues in a nationwide study.
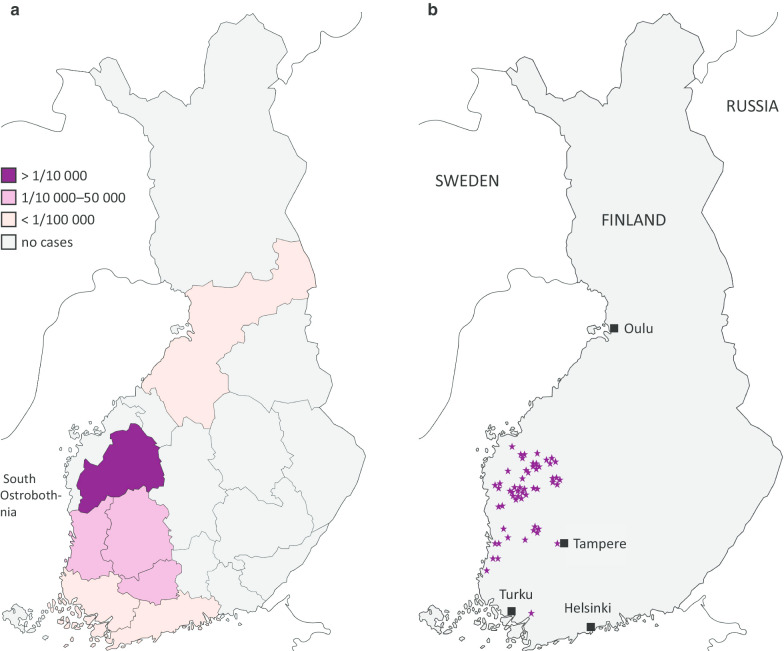
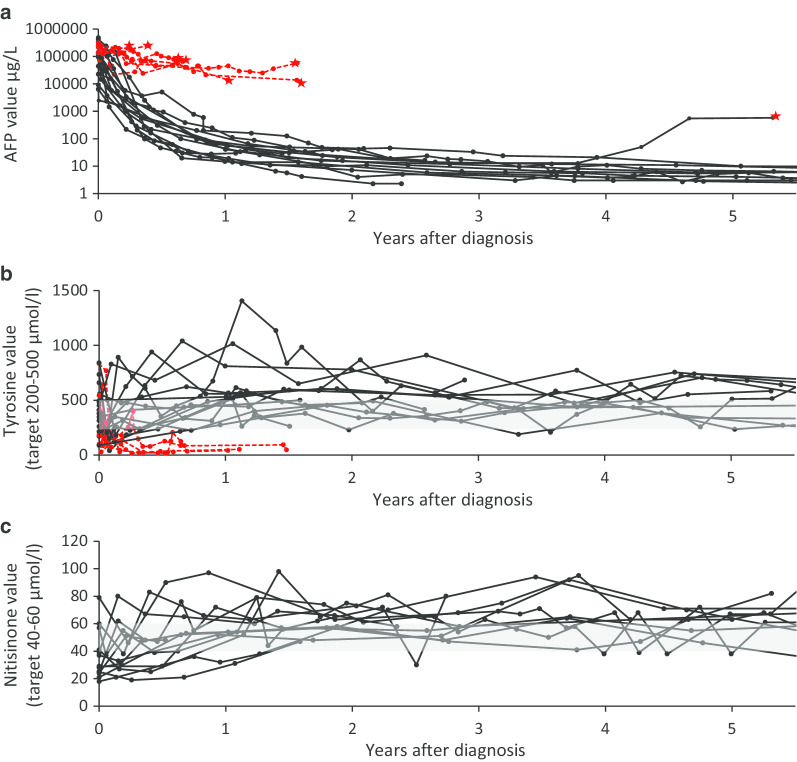
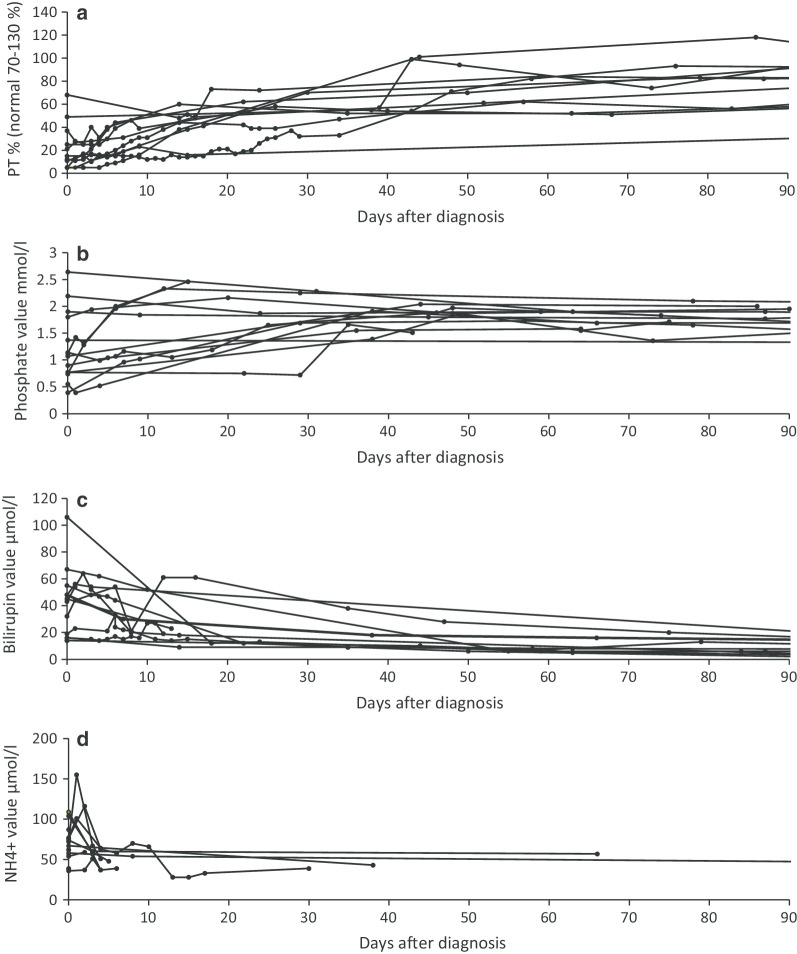
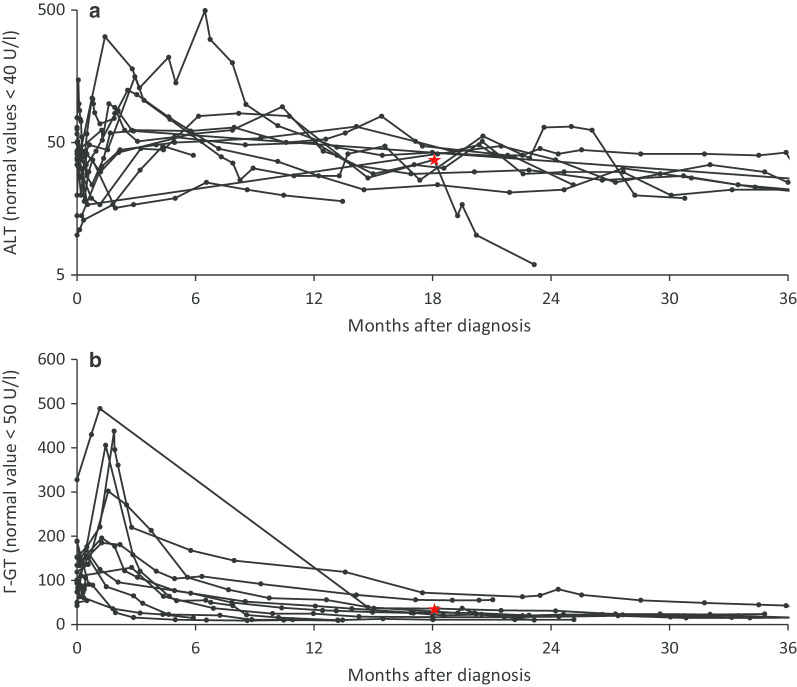
Results: Type 1 tyrosinemia was diagnosed in 22 children in 1978-2019 in Finland. Incidence was 1/90,102, with a significant enrichment in South Ostrobothnia (1/9990). Median age at diagnosis was 5 (range 0.5-36) months, 55% were girls and 13 had homozygotic Trp262X mutation. Four patients were detected through screening and 18 clinically, their main findings being liver failure (50% vs. 100%, respectively, p = 0.026), ascites (0% vs. 53%, p = 0.104), renal tubulopathy (0% vs. 65%, p = 0.035), rickets (25% vs. 65%, p = 0.272), growth failure (0% vs. 66%, p = 0.029), thrombocytopenia (25% vs. 88%, p = 0.028) and anaemia (0% vs. 47%, p = 0.131). One patient was treated with diet, seven with transplantation and 14 with nitisinone. Three late-diagnosed (6-33 months) nitisinone treated patients needed transplantation later. Kidney dysfunction (86% vs. 7%, p = 0.001), hypertension (57% vs. 7%, p = 0.025) and osteopenia/osteoporosis (71% vs. 14%, p = 0.017) were more frequent in transplanted than nitisinone-treated patients. Blood/serum alpha-fetoprotein decreased rapidly on nitisinone in all but one patient, who later developed intrahepatic hepatocellular carcinoma. Liver values normalized in 31 months and other laboratory values except thrombocytopenia within 18 months. Imaging findings normalized in 3-56 months excluding five patients with liver or splenic abnormalities. Low mean nitisinone concentration was associated with higher risk of severe complications (r = 0.758, p = 0.003) despite undetectable urine succinylacetone.
Conclusions: Prognosis of type 1 tyrosinemia has improved in the era of nitisinone, and NBS seems to provide further benefits. Nevertheless, the long-term risk for complications remains, particularly in the case of late diagnosis and/or insufficient nitisinone levels.
Keywords: Liver transplant; Nitisinone; Screening; Succinylacetone; Tyrosinemia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
