

Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions
- PMID: 33046849
- PMCID: PMC7790749
- DOI: 10.1038/s41436-020-00972-3
Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions
Abstract
Purpose: Accurate discrimination of benign and pathogenic rare variation remains a priority for clinical genome interpretation. State-of-the-art machine learning variant prioritization tools are imprecise and ignore important parameters defining gene-disease relationships, e.g., distinct consequences of gain-of-function versus loss-of-function variants. We hypothesized that incorporating disease-specific information would improve tool performance.
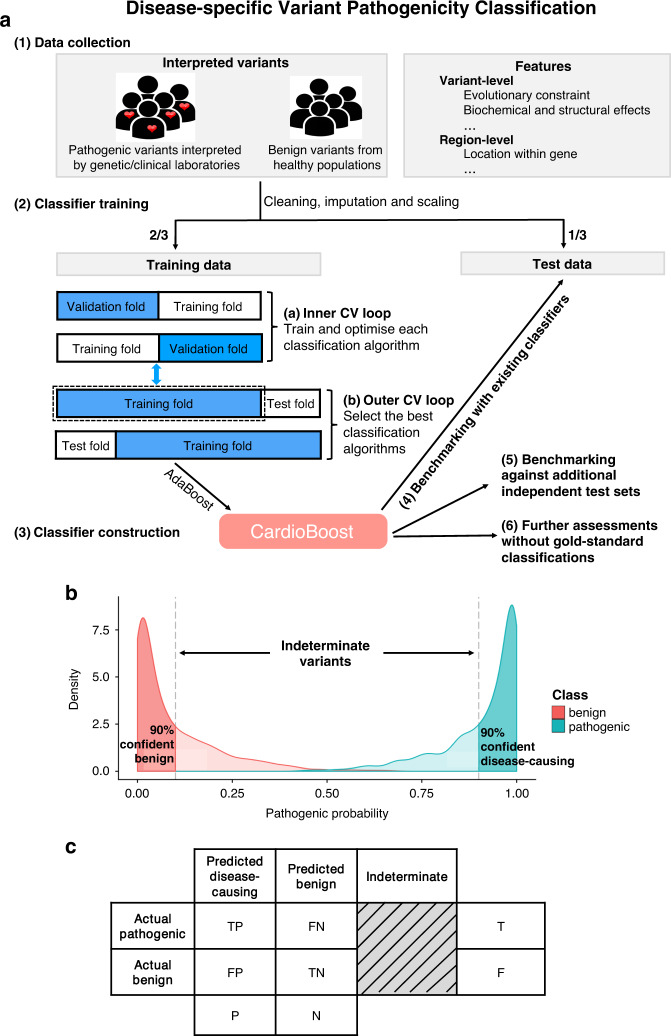
Methods: We developed a disease-specific variant classifier, CardioBoost, that estimates the probability of pathogenicity for rare missense variants in inherited cardiomyopathies and arrhythmias. We assessed CardioBoost's ability to discriminate known pathogenic from benign variants, prioritize disease-associated variants, and stratify patient outcomes.
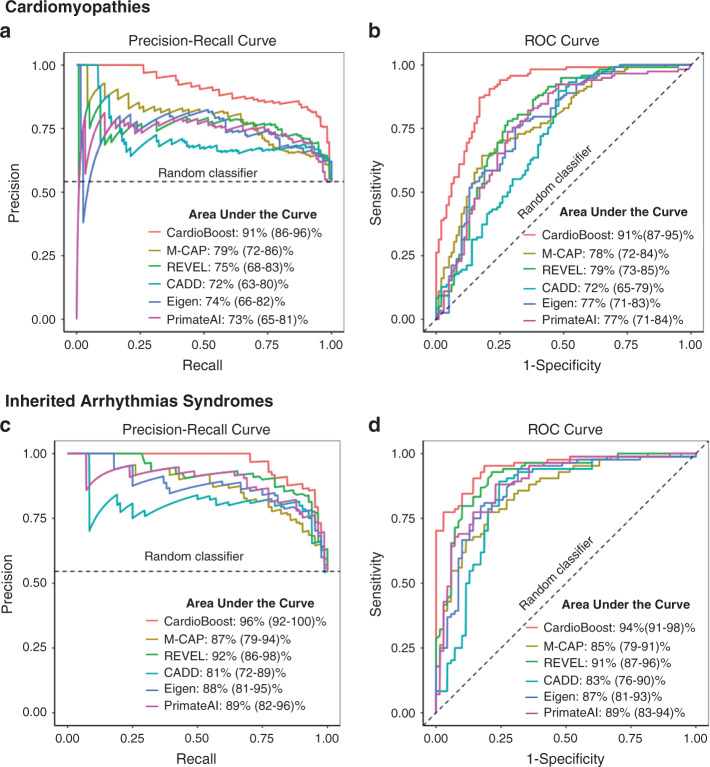
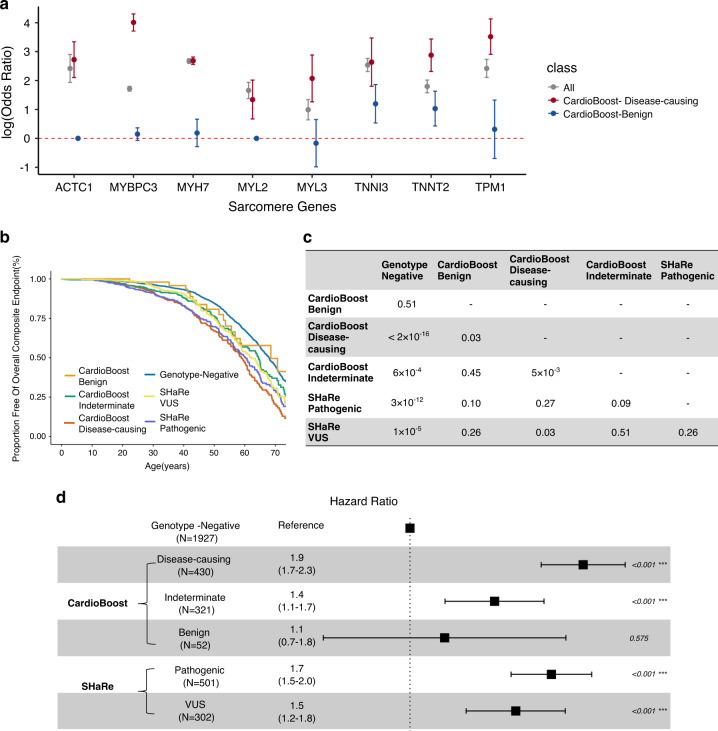
Results: CardioBoost has high global discrimination accuracy (precision recall area under the curve [AUC] 0.91 for cardiomyopathies; 0.96 for arrhythmias), outperforming existing tools (4-24% improvement). CardioBoost obtains excellent accuracy (cardiomyopathies 90.2%; arrhythmias 91.9%) for variants classified with >90% confidence, and increases the proportion of variants classified with high confidence more than twofold compared with existing tools. Variants classified as disease-causing are associated with both disease status and clinical severity, including a 21% increased risk (95% confidence interval [CI] 11-29%) of severe adverse outcomes by age 60 in patients with hypertrophic cardiomyopathy.
Conclusions: A disease-specific variant classifier outperforms state-of-the-art genome-wide tools for rare missense variants in inherited cardiac conditions ( https://www.cardiodb.org/cardioboost/ ), highlighting broad opportunities for improved pathogenicity prediction through disease specificity.
Keywords: Brugada syndrome; cardiomyopathy; long QT syndrome; missense variant interpretation; pathogenicity prediction.
Conflict of interest statement
S.A.C. is a cofounder and director of Enleofen Bio Pte Ltd, a company that develops anti-IL-11 therapeutics. Enleofen Bio had no involvement in this study. J.S.W. and I.O. have consulted for Myokardia, Inc. The ShaRe registry receives research support from MyoKardia. Myokardia had no involvement in this study. The other authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
- MC_UP_1102/19/MRC_/Medical Research Council/United Kingdom
- UL1 TR001863/TR/NCATS NIH HHS/United States
- DH_/Department of Health/United Kingdom
- MC_UP_1605/13/MRC_/Medical Research Council/United Kingdom
- RE/18/4/34215/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- 200990/A/16/Z/WT_/Wellcome Trust/United Kingdom
- NH/17/1/32725/BHF_/British Heart Foundation/United Kingdom
- MC_U120085815/MRC_/Medical Research Council/United Kingdom
- MC_UP_1102/20/MRC_/Medical Research Council/United Kingdom
- 107469/Z/15/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
