

Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy
- PMID: 33046855
- PMCID: PMC8272915
- DOI: 10.1038/s41588-020-0707-1
Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy
Abstract
The influence of genetic background on driver mutations is well established; however, the mechanisms by which the background interacts with Mendelian loci remain unclear. We performed a systematic secondary-variant burden analysis of two independent cohorts of patients with Bardet-Biedl syndrome (BBS) with known recessive biallelic pathogenic mutations in one of 17 BBS genes for each individual. We observed a significant enrichment of trans-acting rare nonsynonymous secondary variants in patients with BBS compared with either population controls or a cohort of individuals with a non-BBS diagnosis and recessive variants in the same gene set. Strikingly, we found a significant over-representation of secondary alleles in chaperonin-encoding genes-a finding corroborated by the observation of epistatic interactions involving this complex in vivo. These data indicate a complex genetic architecture for BBS that informs the biological properties of disease modules and presents a model for secondary-variant burden analysis in recessive disorders.
Conflict of interest statement
Competing interests
N.K. is a founder of, and holds significant stock in, Rescindo Therapeutics.
Figures
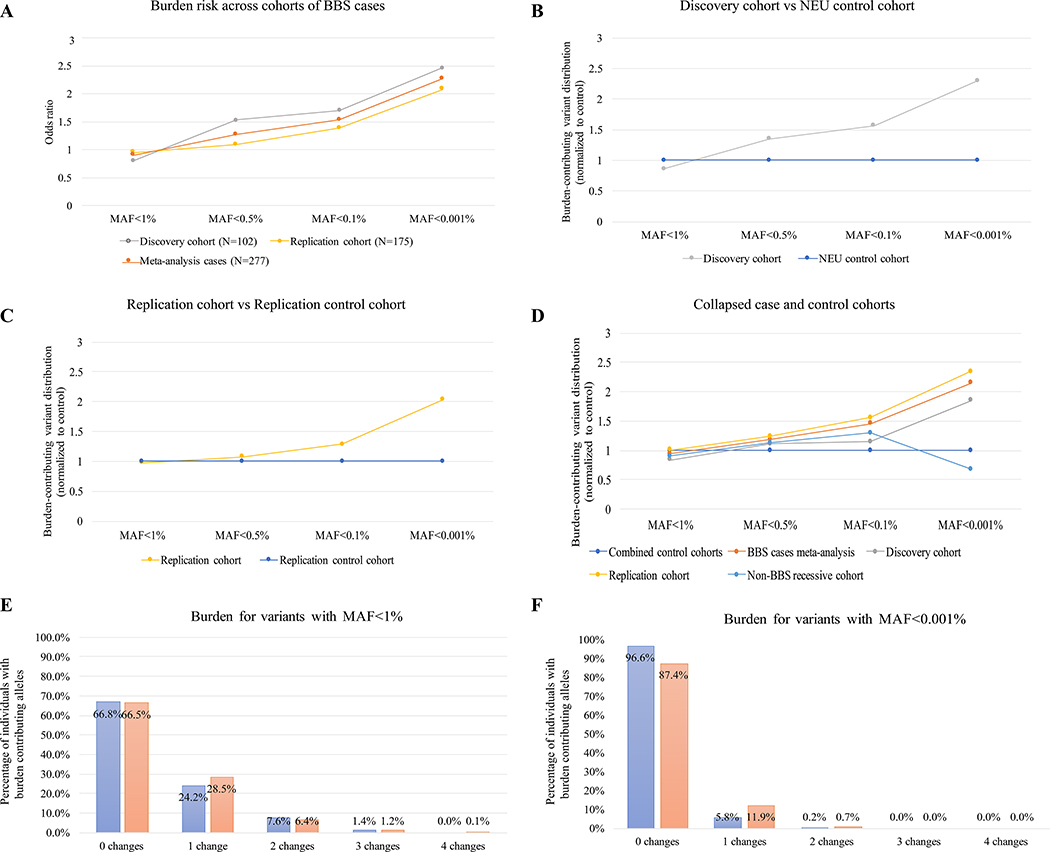

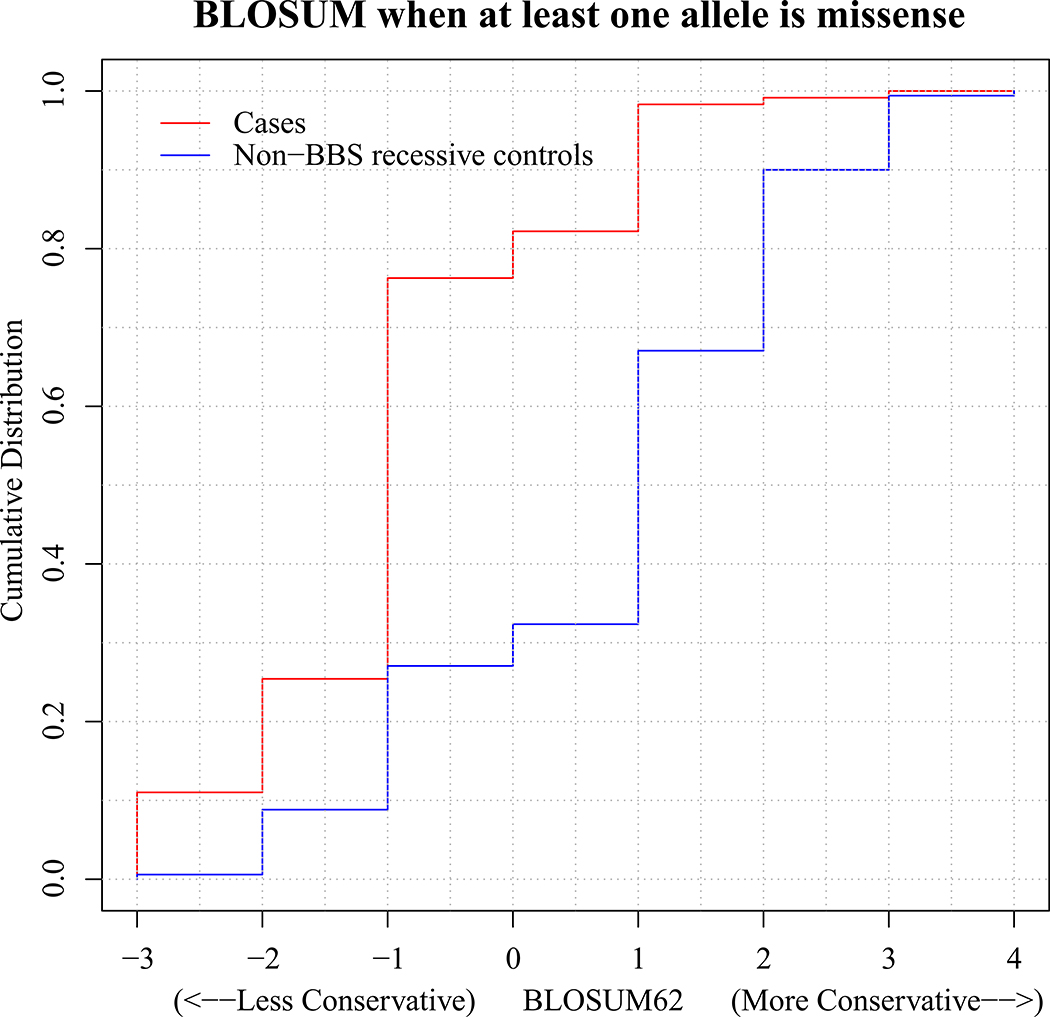
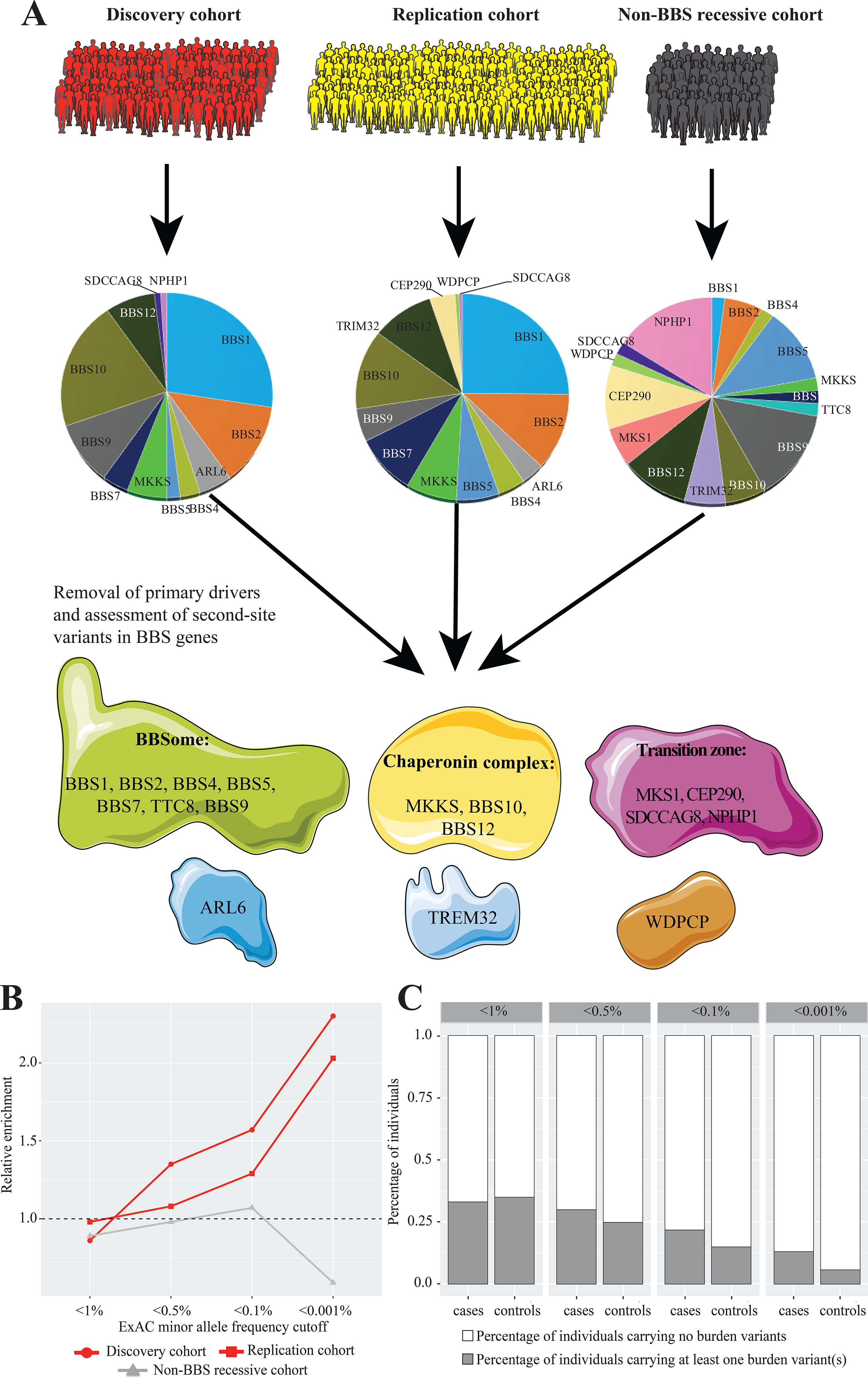
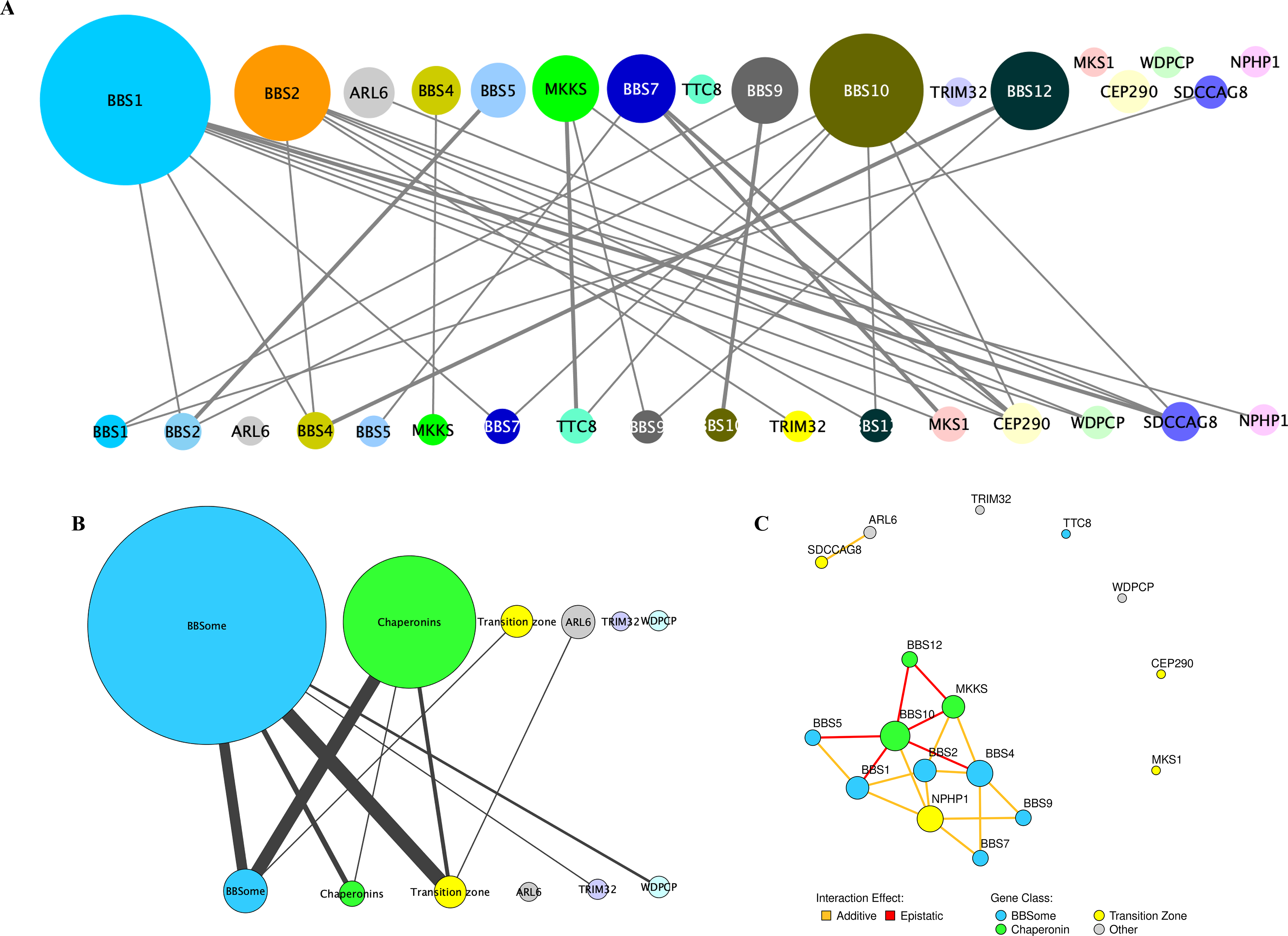
Comment in
-
Secondary-variant genetic burden in Bardet-Biedl syndrome.Nat Rev Nephrol. 2021 Jan;17(1):14. doi: 10.1038/s41581-020-00370-7. Nat Rev Nephrol. 2021. PMID: 33093658 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- R01 MH115957/MH/NIMH NIH HHS/United States
- R01 GM121317/GM/NIGMS NIH HHS/United States
- R01 DK072301/DK/NIDDK NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- U01 HG009080/HG/NHGRI NIH HHS/United States
- T32 GM087237/GM/NIGMS NIH HHS/United States
- R01 DK075972/DK/NIDDK NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- P20 MH084018/MH/NIMH NIH HHS/United States
- R01 HG010372/HG/NHGRI NIH HHS/United States
- R01 HG004037/HG/NHGRI NIH HHS/United States
- U19 CA203654/CA/NCI NIH HHS/United States
- R35 GM127131/GM/NIGMS NIH HHS/United States
- R01 HD081256/HD/NICHD NIH HHS/United States
- R01 HD042601/HD/NICHD NIH HHS/United States
- R01 MH101244/MH/NIMH NIH HHS/United States
- U01 HG009088/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
