

A systematic evaluation of the design and context dependencies of massively parallel reporter assays
- PMID: 33046894
- PMCID: PMC7727316
- DOI: 10.1038/s41592-020-0965-y
A systematic evaluation of the design and context dependencies of massively parallel reporter assays
Abstract
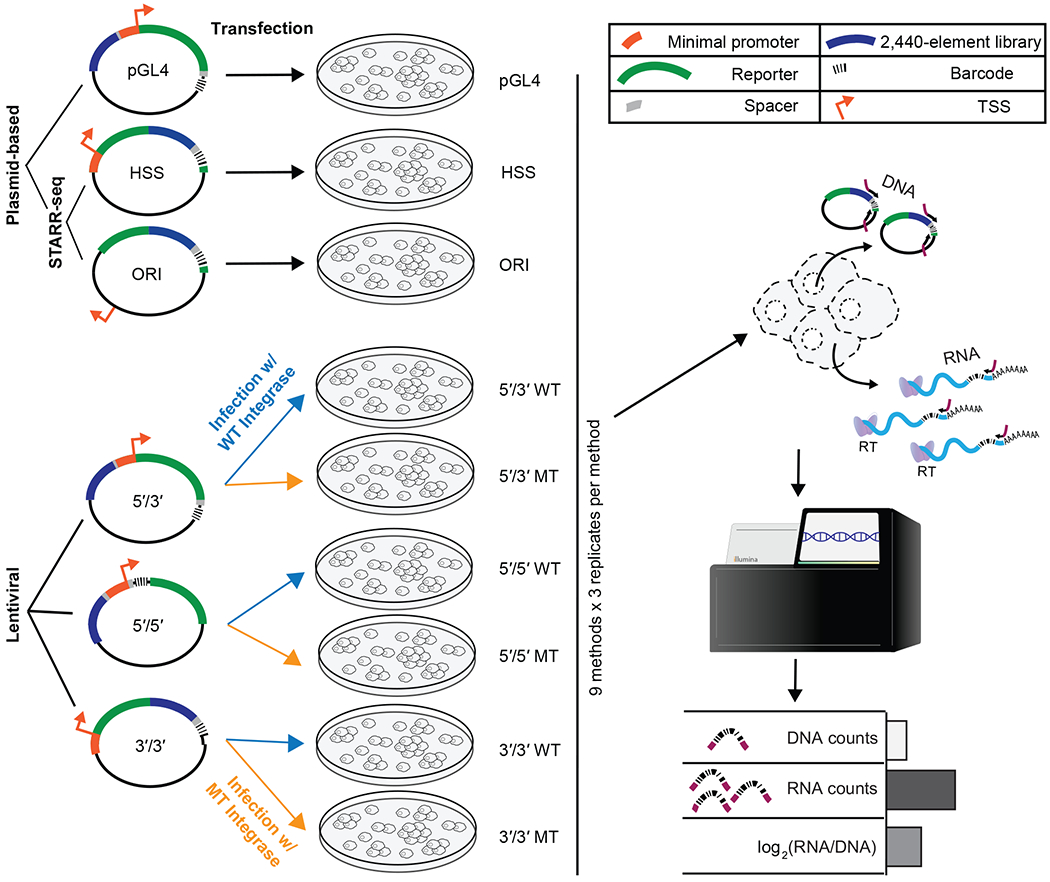
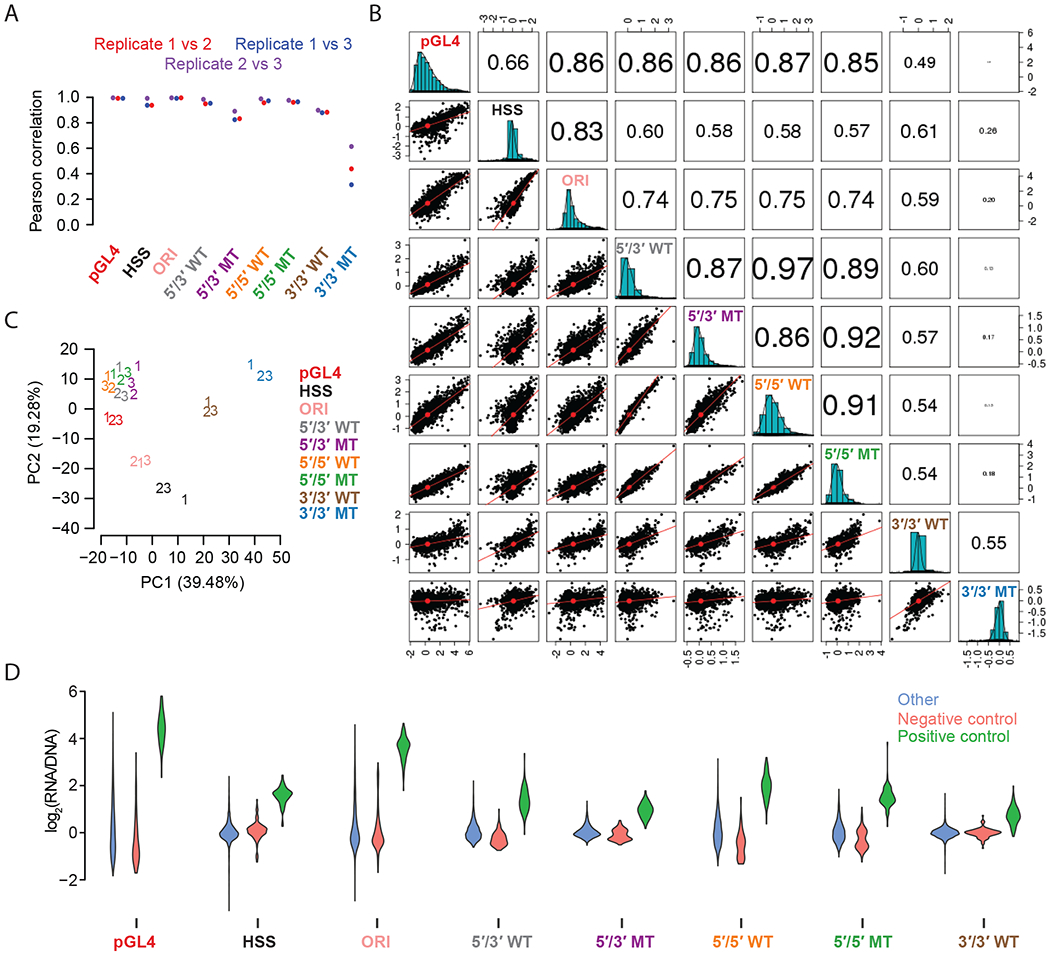
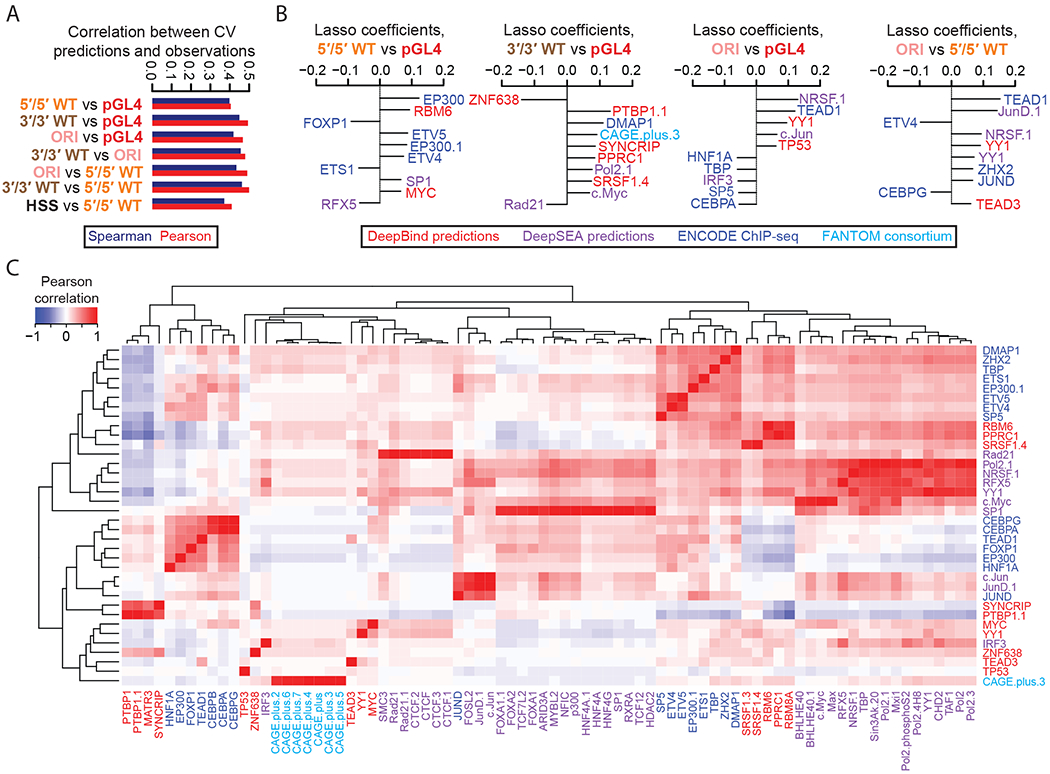
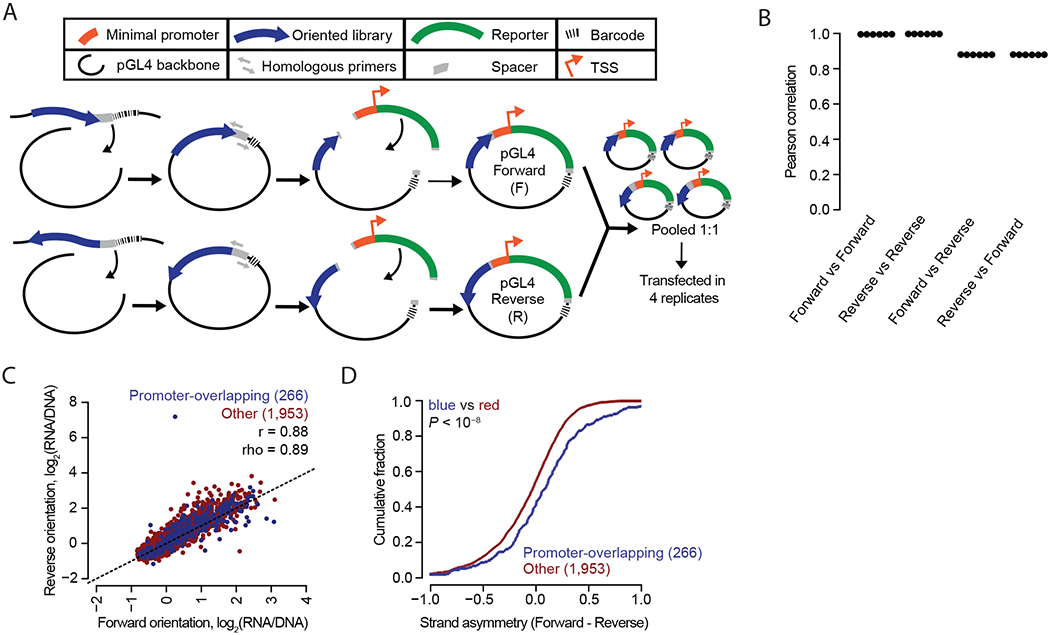
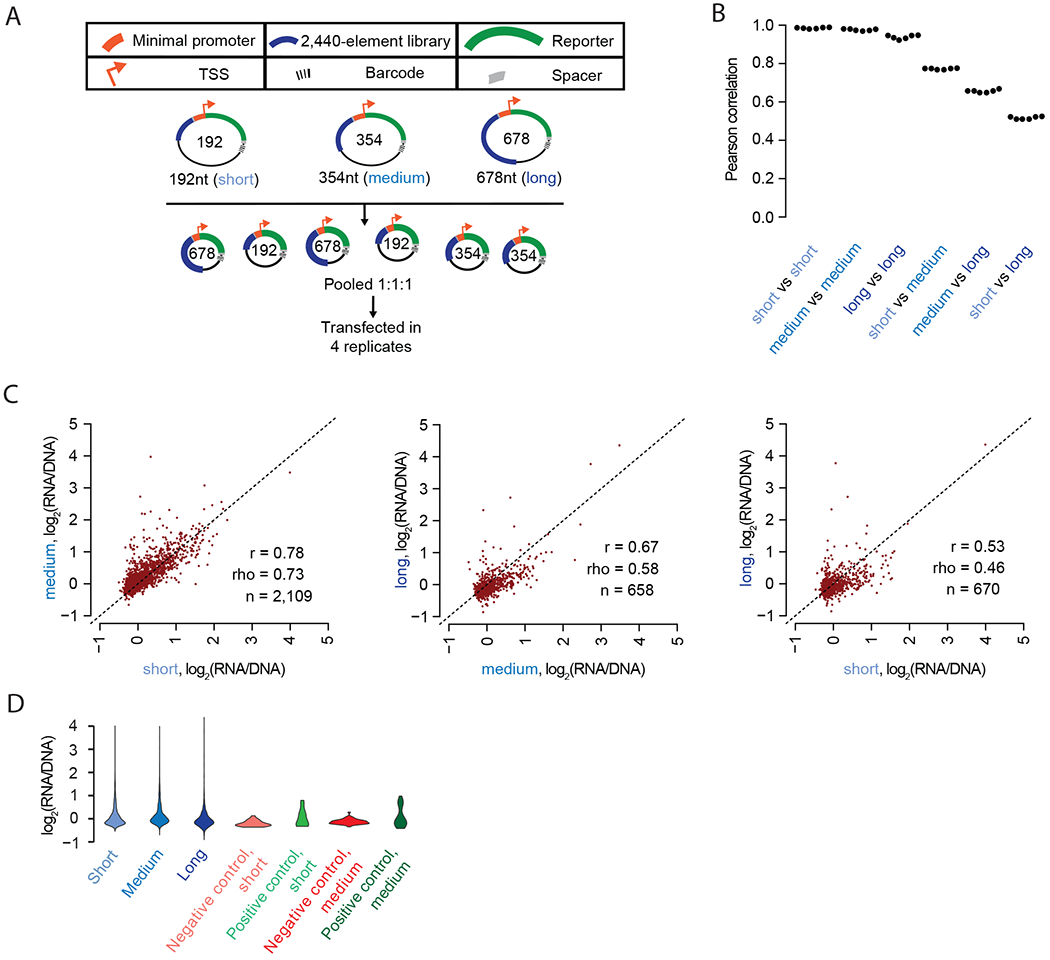
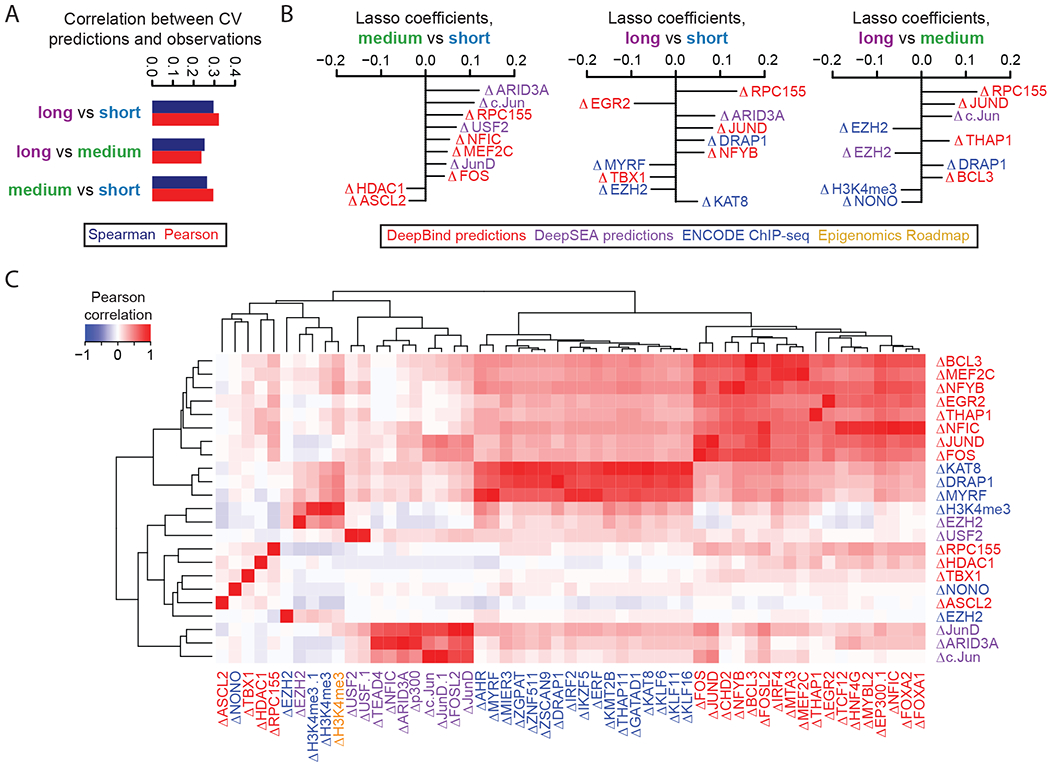
Massively parallel reporter assays (MPRAs) functionally screen thousands of sequences for regulatory activity in parallel. To date, there are limited studies that systematically compare differences in MPRA design. Here, we screen a library of 2,440 candidate liver enhancers and controls for regulatory activity in HepG2 cells using nine different MPRA designs. We identify subtle but significant differences that correlate with epigenetic and sequence-level features, as well as differences in dynamic range and reproducibility. We also validate that enhancer activity is largely independent of orientation, at least for our library and designs. Finally, we assemble and test the same enhancers as 192-mers, 354-mers and 678-mers and observe sizable differences. This work provides a framework for the experimental design of high-throughput reporter assays, suggesting that the extended sequence context of tested elements and to a lesser degree the precise assay, influence MPRA results.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Banerji J, Rusconi S & Schaffner W Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308 (1981). - PubMed
-
- Banerji J, Olson L & Schaffner W A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33, 729–740 (1983). - PubMed
METHODS-ONLY REFERENCES
-
- Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN] (2013).
Publication types
MeSH terms
Substances
Grants and funding
- U01 MH116438/MH/NIMH NIH HHS/United States
- T32 HL007093/HL/NHLBI NIH HHS/United States
- UM1 HG009408/HG/NHGRI NIH HHS/United States
- R01 MH109907/MH/NIMH NIH HHS/United States
- F30 HG009479/HG/NHGRI NIH HHS/United States
- P30 DK098722/DK/NIDDK NIH HHS/United States
- R21 HG010065/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R21 HG010683/HG/NHGRI NIH HHS/United States
- M01 RR001271/RR/NCRR NIH HHS/United States
- R01 HG009136/HG/NHGRI NIH HHS/United States
- R01 MH116438/MH/NIMH NIH HHS/United States
- R01 HG006768/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
