

Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms
- PMID: 33050168
- PMCID: PMC7599937
- DOI: 10.3390/cancers12102891
Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms
Abstract
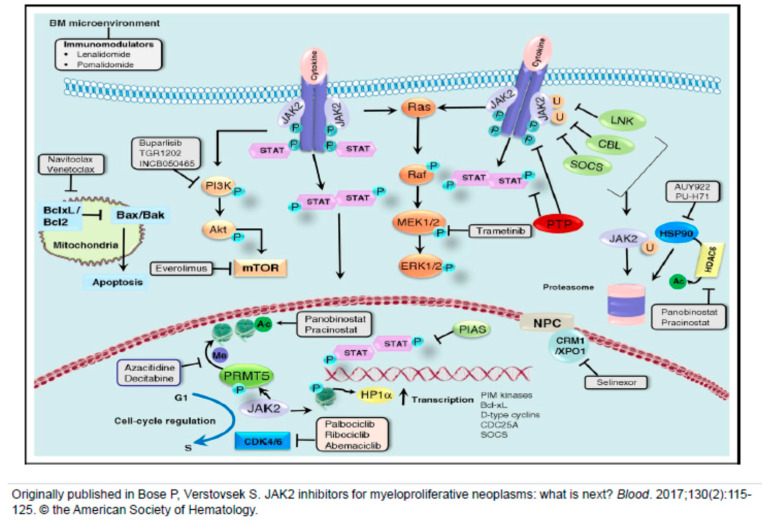
Janus kinase (JAK) inhibition forms the cornerstone of the treatment of myelofibrosis (MF), and the JAK inhibitor ruxolitinib is often used as a second-line agent in patients with polycythemia vera (PV) who fail hydroxyurea (HU). In addition, ruxolitinib continues to be studied in patients with essential thrombocythemia (ET). The benefits of JAK inhibition in terms of splenomegaly and symptoms in patients with MF are undeniable, and ruxolitinib prolongs the survival of persons with higher risk MF. Despite this, however, "disease-modifying" effects of JAK inhibitors in MF, i.e., bone marrow fibrosis and mutant allele burden reduction, are limited. Similarly, in HU-resistant/intolerant PV, while ruxolitinib provides excellent control of the hematocrit, symptoms and splenomegaly, reduction in the rate of thromboembolic events has not been convincingly demonstrated. Furthermore, JAK inhibitors do not prevent disease evolution to MF or acute myeloid leukemia (AML). Frontline cytoreductive therapy for PV generally comprises HU and interferons, which have their own limitations. Numerous novel agents, representing diverse mechanisms of action, are in development for the treatment of these three classic myeloproliferative neoplasms (MPNs). JAK inhibitor-based combinations, all of which are currently under study for MF, have been covered elsewhere in this issue. In this article, we focus on agents that have been studied as monotherapy in patients with MF, generally after JAK inhibitor resistance/intolerance, as well as several novel compounds in development for PV/ET.
Keywords: CPI-0610; KRT-232; PRM-151; imetelstat; ropeginterferon alfa-2b.
Conflict of interest statement
P.B. reports research funding from Incyte, Celgene (now BMS), CTI BioPharma, Constellation Pharmaceuticals, Kartos Therapeutics, Blueprint Medicines, Astellas, Pfizer, NS Pharma and Promedior, and honoraria from Incyte, Celgene (now BMS), CTI BioPharma, Kartos Therapeutics, and Blueprint Medicines. S.V. reports research support from Incyte, Roche, NS Pharma, Celgene (now BMS), Gilead, Promedior, CTI BioPharma, Genentech, Blueprint Medicines, Novartis, Sierra Oncology, Pharma Essentia, Astra Zeneca, Ital Pharma and Protagonist Therapeutics, and consulting fees from Constellation Pharmaceuticals, Pragmatist, Sierra Oncology, Incyte, Novartis and Celgene (now BMS). L.M. declares no competing conflicts of interest.
Figures

References
-
- Rampal R., Al-Shahrour F., Abdel-Wahab O., Patel J.P., Brunel J.P., Mermel C.H., Bass A.J., Pretz J., Ahn J., Hricik T., et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123:123–133. doi: 10.1182/blood-2014-02-554634. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
