

Clinical Genetics Can Solve the Pitfalls of Genome-Wide Investigations: Lesson from Mismapping a Loss-of-Function Variant in KANSL1
- PMID: 33050294
- PMCID: PMC7600039
- DOI: 10.3390/genes11101177
Clinical Genetics Can Solve the Pitfalls of Genome-Wide Investigations: Lesson from Mismapping a Loss-of-Function Variant in KANSL1
Abstract
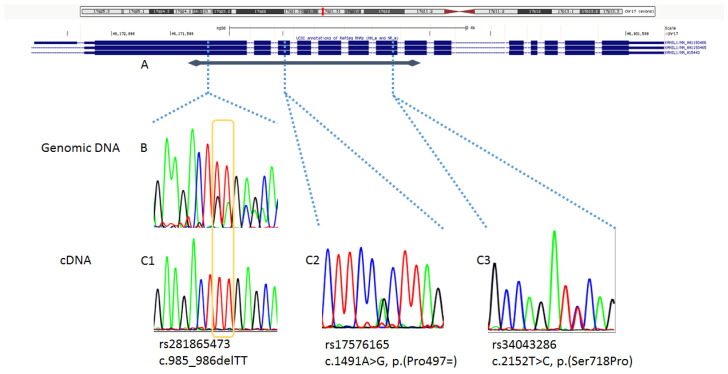
Massive parallel sequencing of 70 genes in a girl with a suspicion of chromatinopathy detected the (NM_015443.4:)c.985_986delTT variant in exon 2 of KANSL1, which led to a diagnostic consideration of Koolen De Vries syndrome. The same variant was present in the healthy mother, consistent with either incomplete penetrance or variant mismapping. A network of second opinion was implemented among clinical geneticists first, and a diagnosis of Koolen De Vries syndrome was considered unlikely. By MLPA, a duplication spanning exons 1-3 of KANSL1 was detected in both the mother and the daughter. On cDNA sequencing, biallelic wild type mRNA was observed. We concluded that the variant affects the noncoding duplicated gene region in our family, and we finally classified it as benign. Parallel wide genomic sequencing is increasingly the first genetic investigation in individuals with intellectual disability. The c.985_986delTT variant in KANSL1 was described both in individuals with typical KdVS and in a limited number of healthy subjects. This report highlights the role of clinical genetics to correctly classify variants and to define proper clinical and diagnostic correlations.
Keywords: KANSL1; clinical evaluation; copy number polymorphisms; variant interpretation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Shaw-Smith C., Pittman A.M., Willatt L., Martin H., Rickman L., Gribble S., Curley R., Cumming S., Dunn C., Kalaitzopoulos D., et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. - DOI - PubMed
-
- Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C., et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
