

Stearic Acid and TNF-α Co-Operatively Potentiate MIP-1α Production in Monocytic Cells via MyD88 Independent TLR4/TBK/IRF3 Signaling Pathway
- PMID: 33050324
- PMCID: PMC7600458
- DOI: 10.3390/biomedicines8100403
Stearic Acid and TNF-α Co-Operatively Potentiate MIP-1α Production in Monocytic Cells via MyD88 Independent TLR4/TBK/IRF3 Signaling Pathway
Abstract
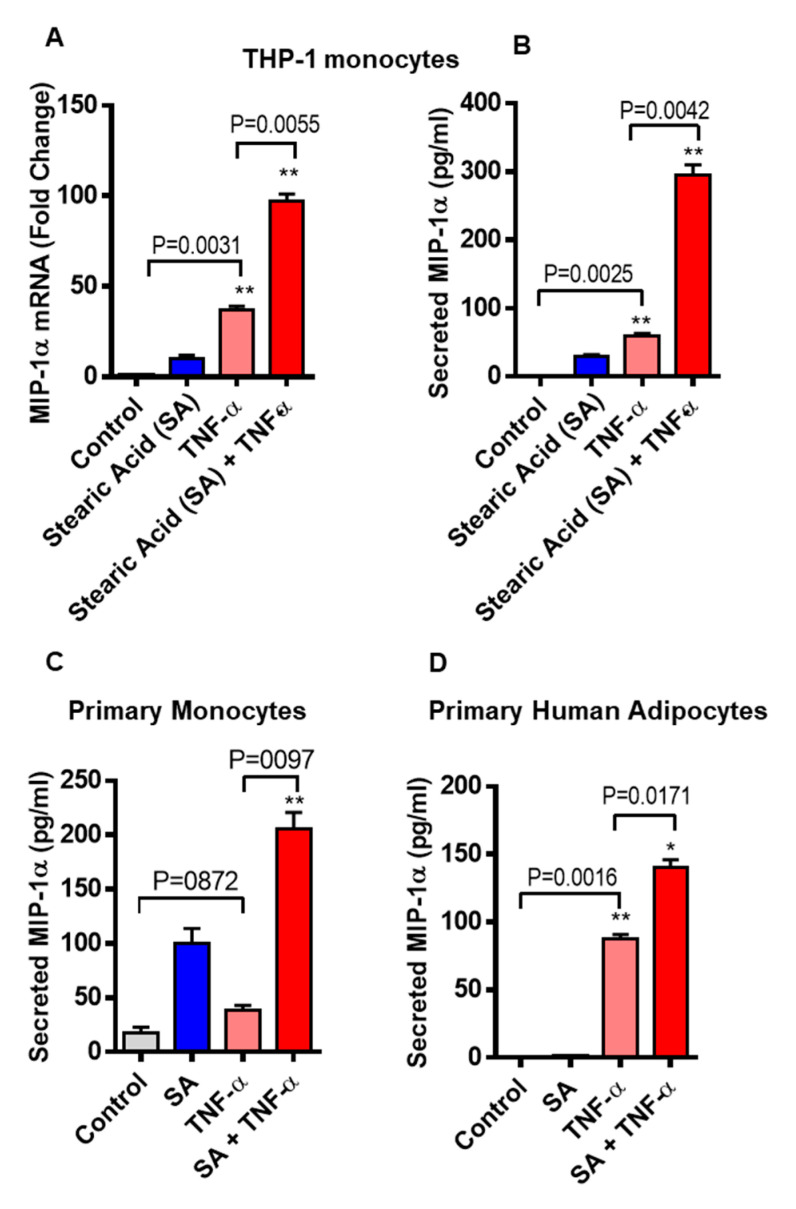
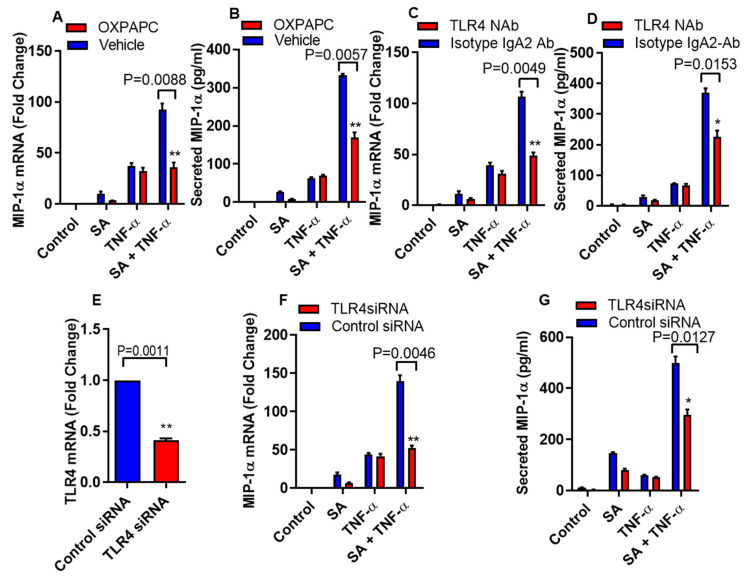
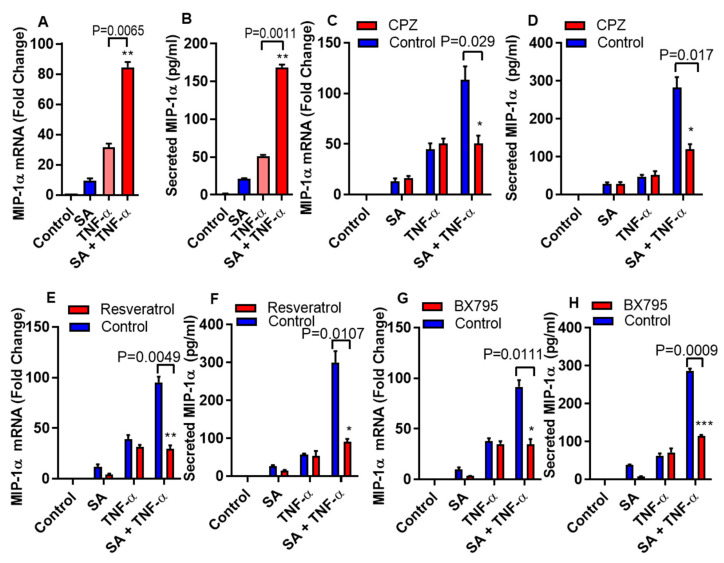
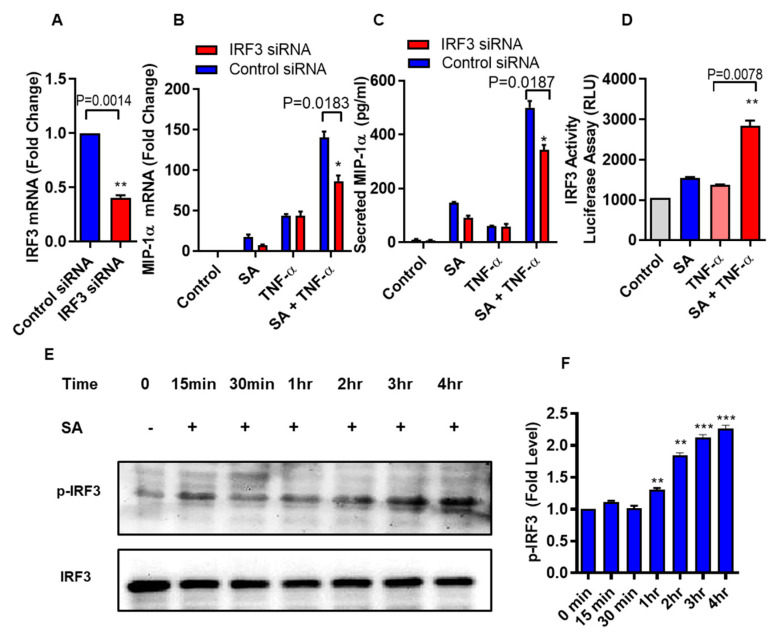
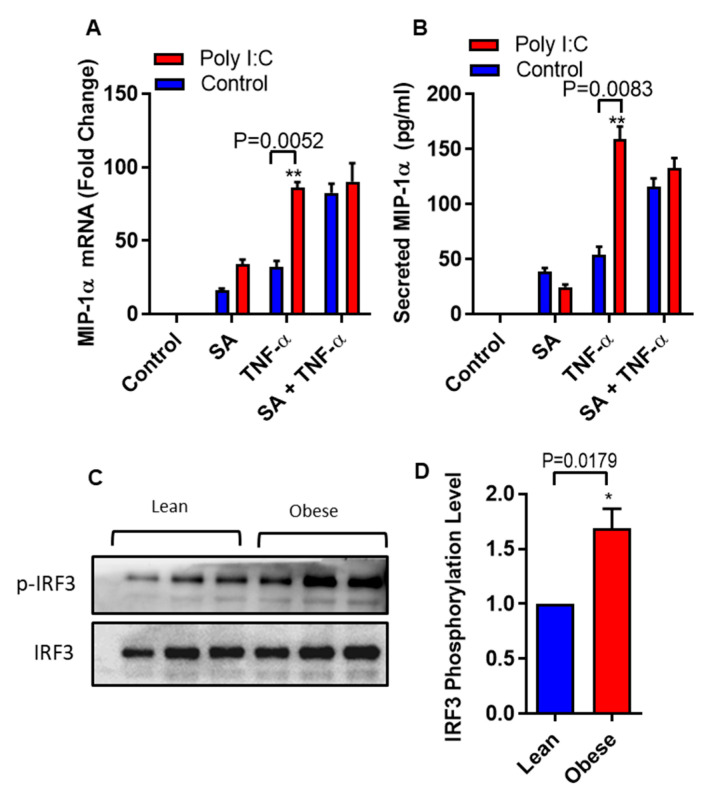
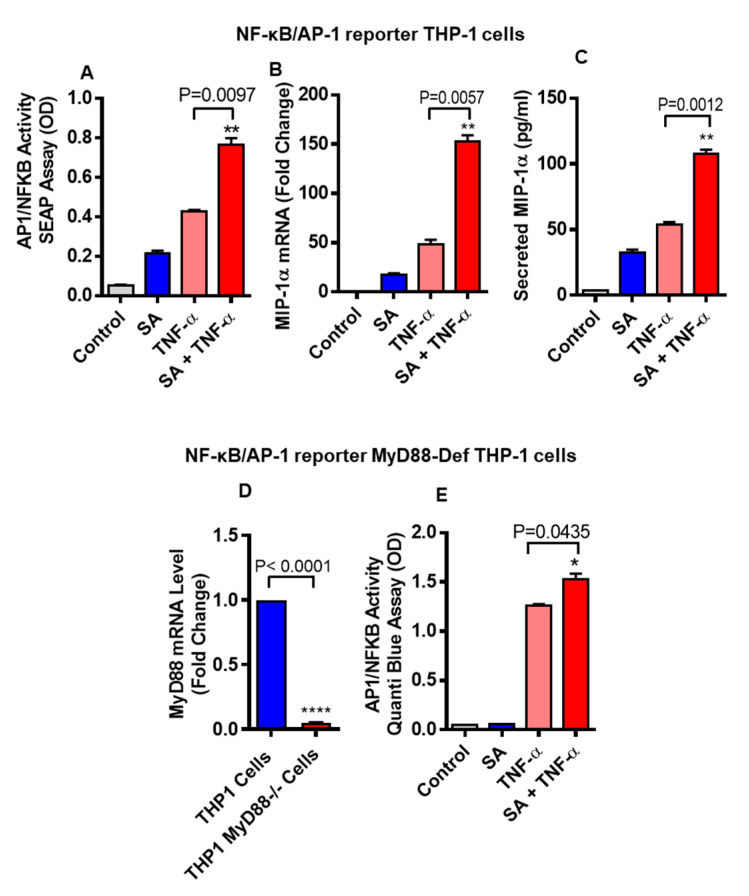
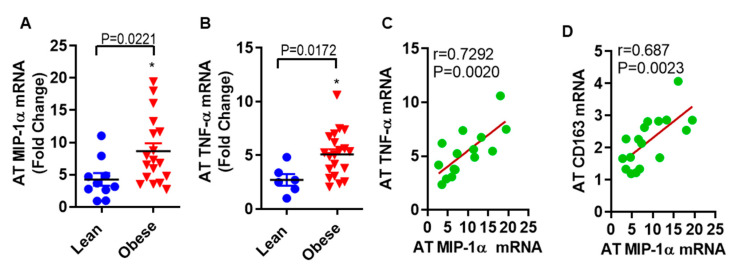
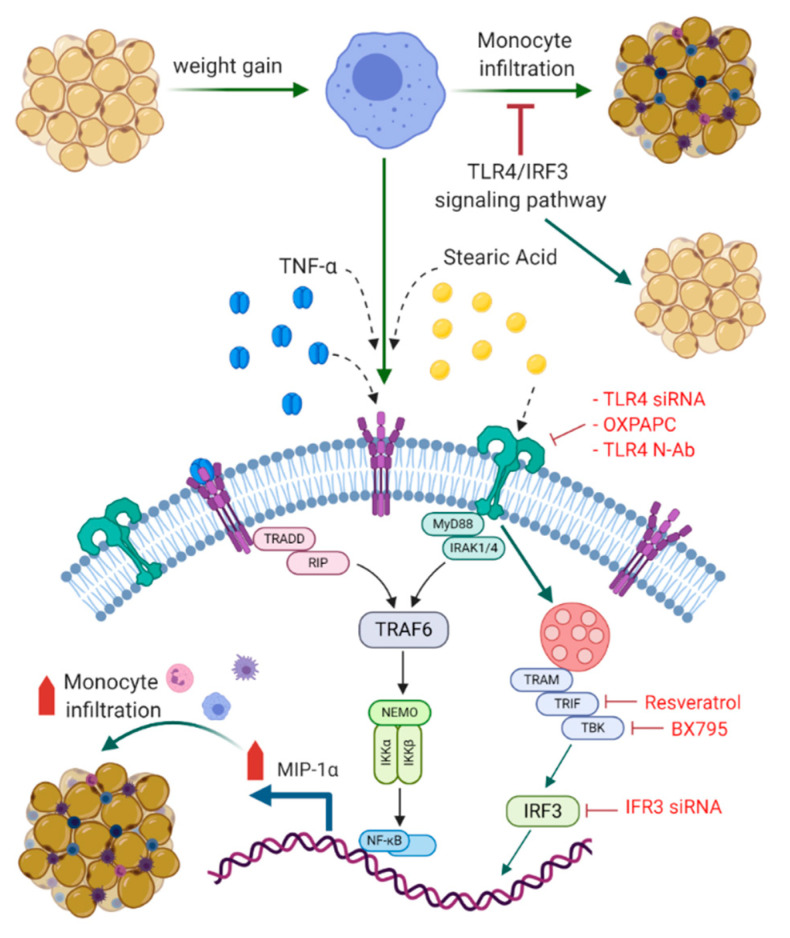
Increased circulatory and adipose tissue expression of macrophage inflammatory protein (MIP)-1α (CC motif chemokine ligand-3/CCL3) and its association with inflammation in the state of obesity is well documented. Since obesity is associated with increases in both stearic acid and tumor necrosis factor α (TNF-α) in circulation, we investigated whether stearic acid and TNF-α together could regulate MIP-1α/CCL3 expression in human monocytic cells, and if so, which signaling pathways were involved in MIP-1α/CCL3 modulation. Monocytic cells were treated with stearic acid and TNF-α resulted in enhanced production of MIP-1α/CCL3 compared to stearic acid or TNF-α alone. To explore the underlying mechanisms, cooperative effect of stearic acid for MIP-α/CCL3 expression was reduced by TLR4 blocking, and unexpectedly we found that the synergistic production of MIP-α/CCL3 in MyD88 knockout (KO) cells was not suppressed. In contrast, this MIP-α/CCL3 expression was attenuated by inhibiting TBK1/IRF3 activity. Cells deficient in IRF3 did not show cooperative effect of stearate/TNF-α on MIP-1α/CCL3 production. Furthermore, activation of IRF3 by polyinosinic-polycytidylic acid (poly I:C) produced a cooperative effect with TNF-α for MIP-1α/CCL3 production that was comparable to stearic acid. Individuals with obesity show high IRF3 expression in monocytes as compared to lean individuals. Furthermore, elevated levels of MIP-1α/CCL3 positively correlate with TNF-α and CD163 in fat tissues from individuals with obesity. Taken together, this study provides a novel model for the pathologic role of stearic acid to produce MIP-1α/CCL3 in the presence of TNF-α associated with obesity settings.
Keywords: IRF3; MIP-1α; TLR4; TNF-α/CCL3; TRIF/TBK; obesity; stearic acid.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Stephens J.M., Pekala P.H. Transcriptional repression of the c/ebp-alpha and glut4 genes in 3t3-l1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J. Biol. Chem. 1992;267:13580–13584. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
