

The Dual Role of Glutamatergic Neurotransmission in Alzheimer's Disease: From Pathophysiology to Pharmacotherapy
- PMID: 33050345
- PMCID: PMC7589203
- DOI: 10.3390/ijms21207452
The Dual Role of Glutamatergic Neurotransmission in Alzheimer's Disease: From Pathophysiology to Pharmacotherapy
Abstract
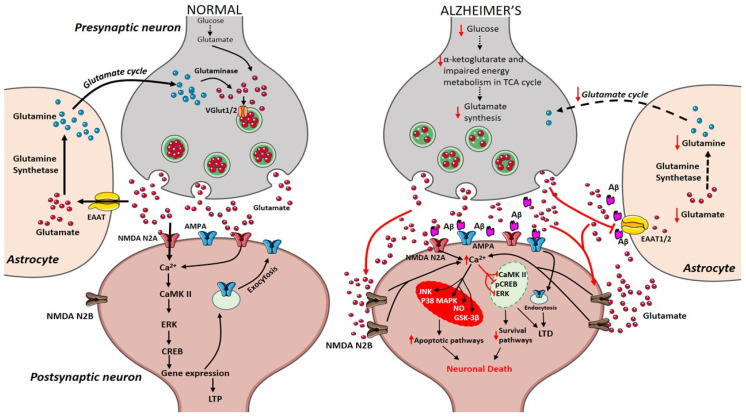
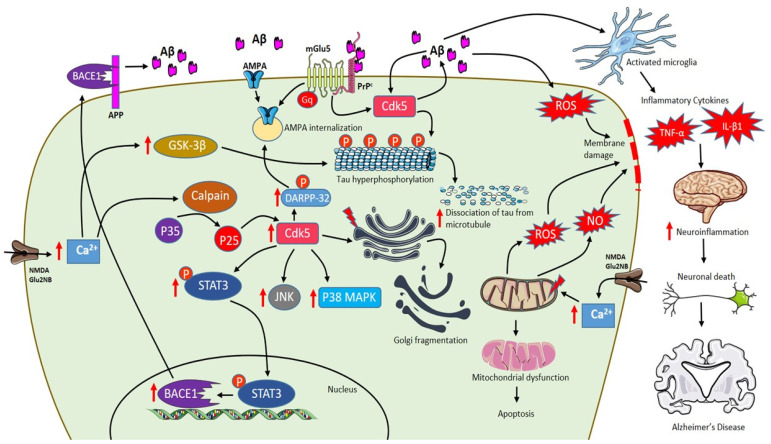
Alzheimer's disease (AD) is an age-related dementia and neurodegenerative disorder, characterized by Aβ and tau protein deposition impairing learning, memory and suppressing synaptic plasticity of neurons. Increasing evidence suggests that there is a link between the glucose and glutamate alterations with age that down-regulates glucose utilization reducing glutamate levels in AD patients. Deviations in brain energy metabolism reinforce the development of AD by hampering glutamate levels in the brain. Glutamate is a nonessential amino acid and the major excitatory neurotransmitter synthesized from glucose. Alterations in cerebral glucose and glutamate levels precede the deposition of Aβ plaques. In the brain, over 40% of neuronal synapses are glutamatergic and disturbances in glutamatergic function have been implicated in pathophysiology of AD. Nevertheless, targeting the glutamatergic system seems to be a promising strategy to develop novel, improved therapeutics for AD. Here, we review data supporting the involvement of the glutamatergic system in AD pathophysiology as well as the efficacy of glutamatergic agents in this neurodegenerative disorder. We also discuss exciting new prospects for the development of improved therapeutics for this devastating disorder.
Keywords: AD; AMPA; EAAT1/2; NMDA; ageing; amyoid-β; glucose; glutamate; metabotropic receptors; tau; therapeutic targets.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Coronel R., Palmer C., Bernabeu-Zornoza A., Monteagudo M., Rosca A., Zambrano A., Liste I. Physiological effects of amyloid precursor protein and its derivatives on neural stem cell biology and signaling pathways involved. Neural Regen. Res. 2019;14:1661–1671. doi: 10.4103/1673-5374.257511. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
