

Release from cell cycle arrest with Cdk4/6 inhibitors generates highly synchronized cell cycle progression in human cell culture
- PMID: 33052073
- PMCID: PMC7653349
- DOI: 10.1098/rsob.200200
Release from cell cycle arrest with Cdk4/6 inhibitors generates highly synchronized cell cycle progression in human cell culture
Abstract
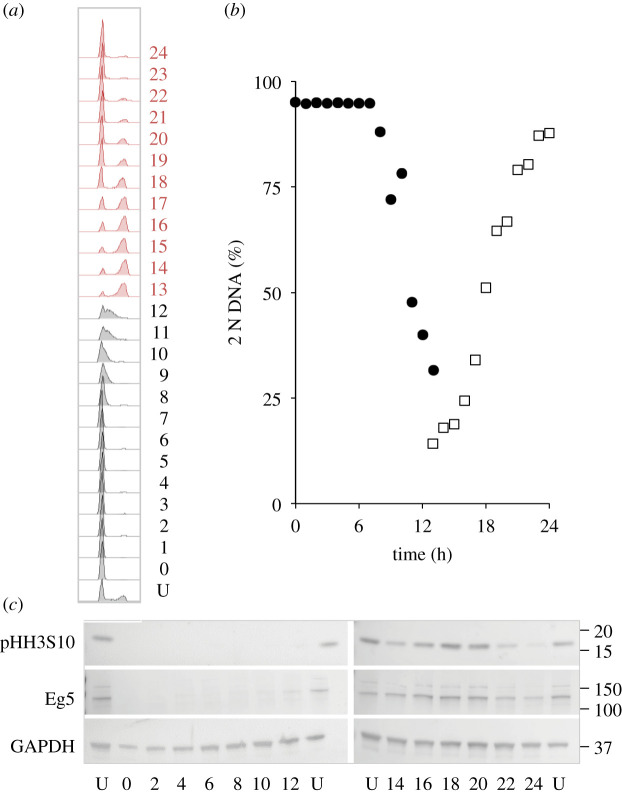
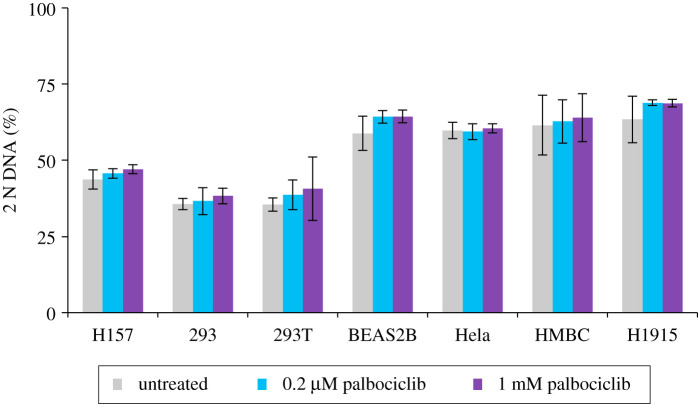
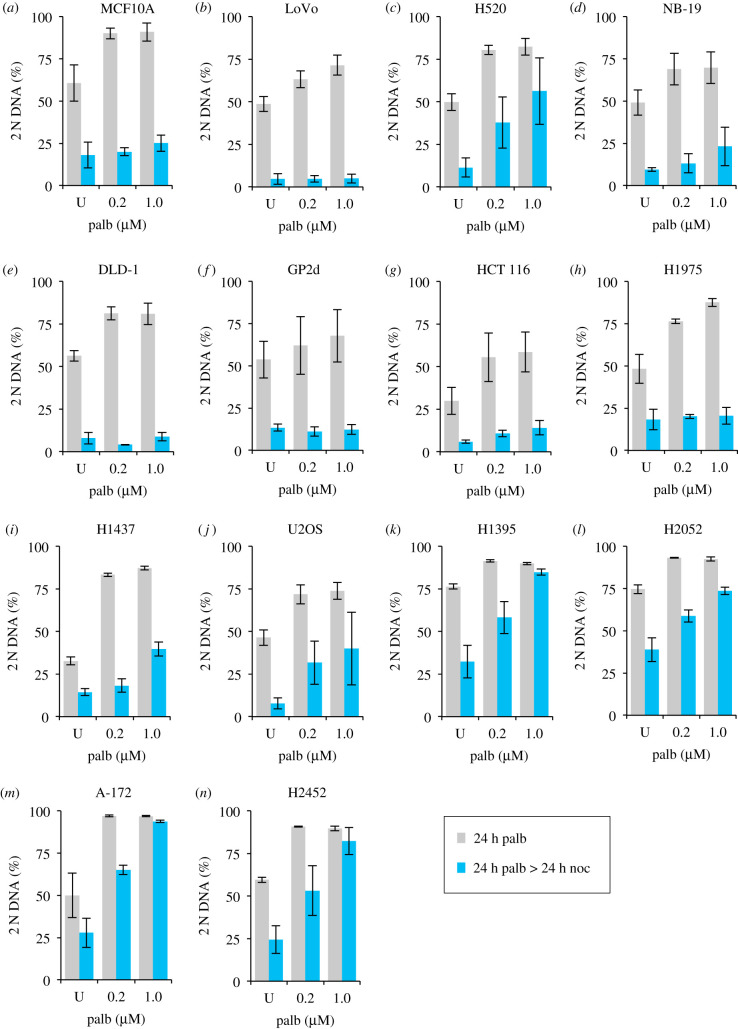
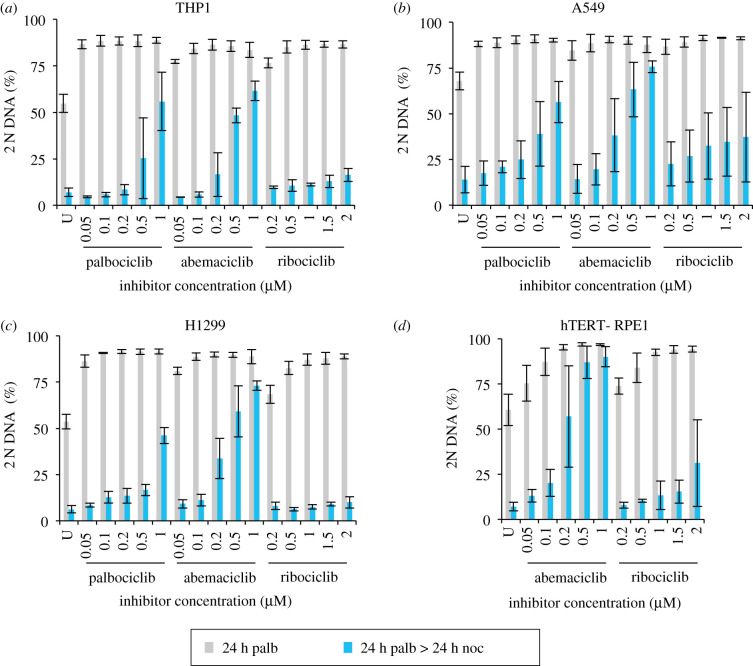
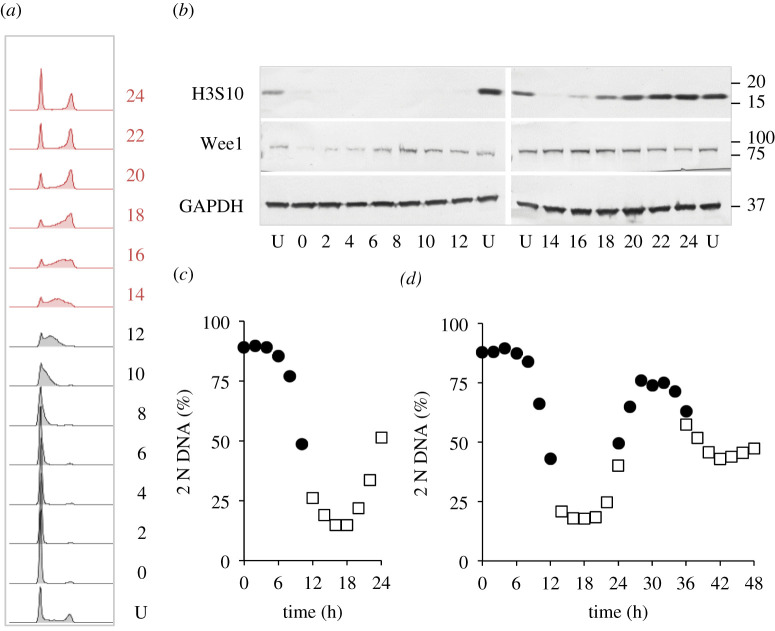
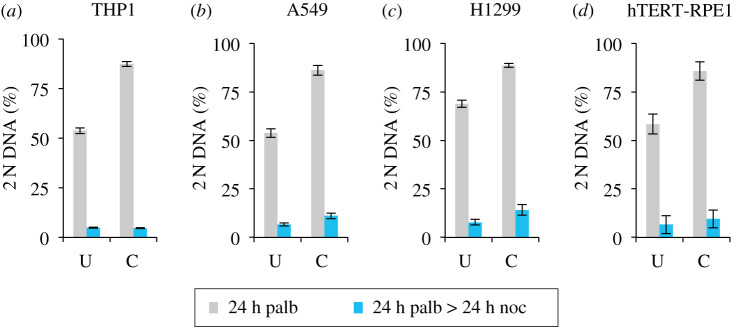
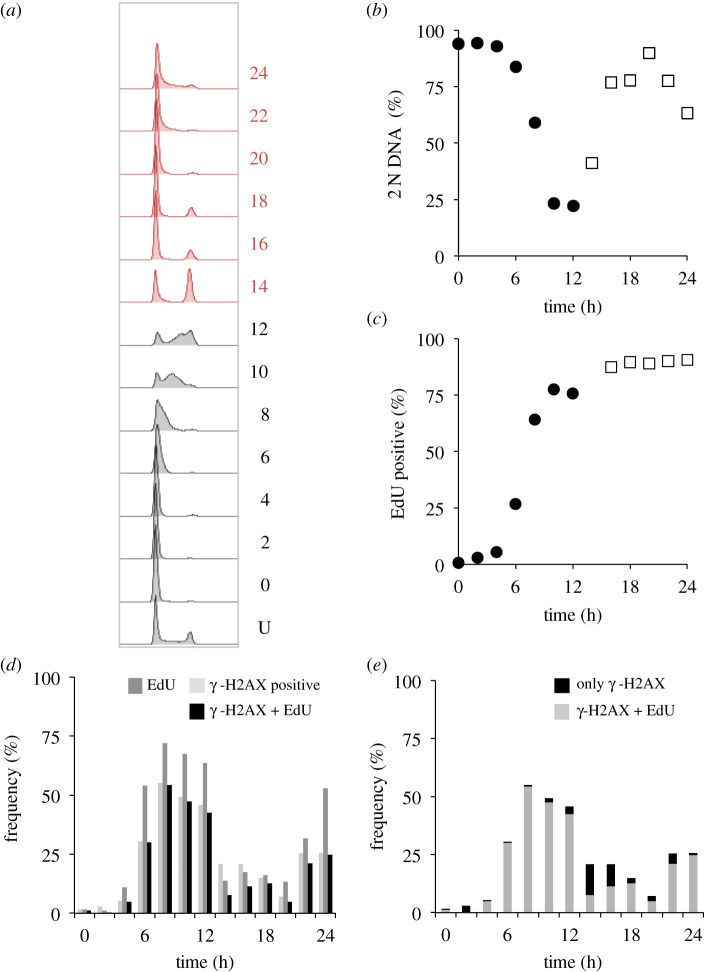
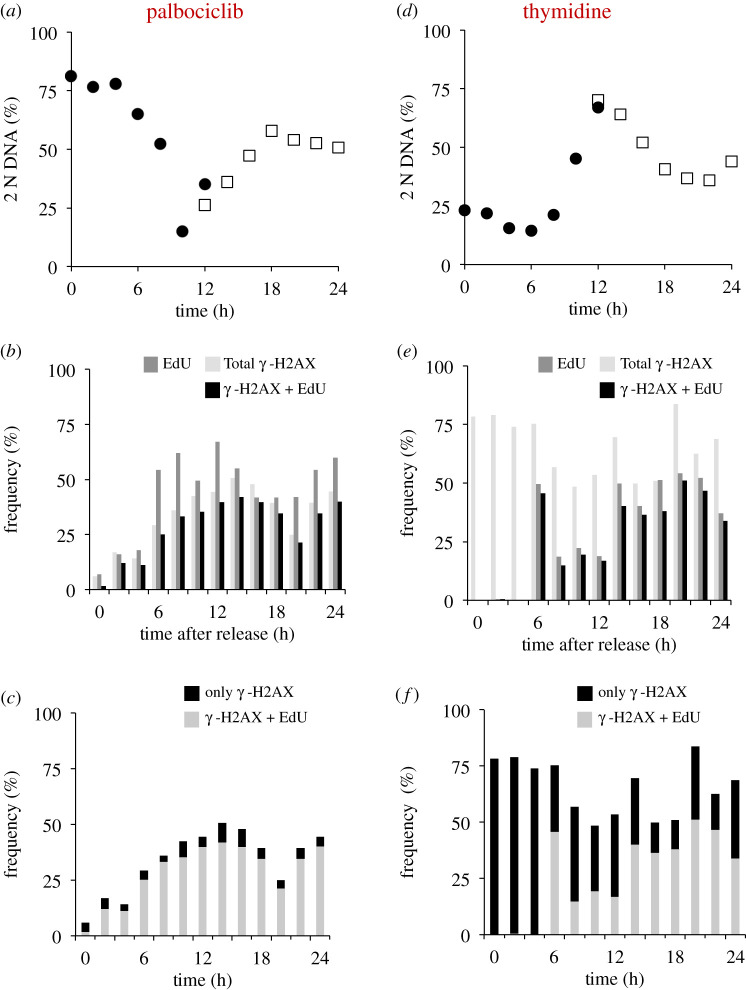
Each approach used to synchronize cell cycle progression of human cell lines presents a unique set of challenges. Induction synchrony with agents that transiently block progression through key cell cycle stages are popular, but change stoichiometries of cell cycle regulators, invoke compensatory changes in growth rate and, for DNA replication inhibitors, damage DNA. The production, replacement or manipulation of a target molecule must be exceptionally rapid if the interpretation of phenotypes in the cycle under study is to remain independent of impacts upon progression through the preceding cycle. We show how these challenges are avoided by exploiting the ability of the Cdk4/6 inhibitors, palbociclib, ribociclib and abemaciclib to arrest cell cycle progression at the natural control point for cell cycle commitment: the restriction point. After previous work found no change in the coupling of growth and division during recovery from CDK4/6 inhibition, we find high degrees of synchrony in cell cycle progression. Although we validate CDK4/6 induction synchronization with hTERT-RPE-1, A549, THP1 and H1299, it is effective in other lines and avoids the DNA damage that accompanies synchronization by thymidine block/release. Competence to return to cycle after 72 h arrest enables out of cycle target induction/manipulation, without impacting upon preceding cycles.
Keywords: CDK4; cell cycle; cell cycle synchronization; palbociclib; restriction point; thymidine block.
Conflict of interest statement
We have no competing interests to declare.
Figures



References
-
- Mitchison JM. 1971. Synchronous cultures. In The biology of the cell cycle (ed. Mitchison JM.), pp. 25–57. London, NY: Cambridge University Press.
-
- Banfalvi G. 2011. Overview of cell synchronization. In Cell cycle synchronization (ed. Banfalvi G.), pp. 1–23. Totowa, NJ: Humana Press.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources