

Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies
- PMID: 33053333
- PMCID: PMC7562821
- DOI: 10.1016/j.immuni.2020.09.015
Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies
Abstract
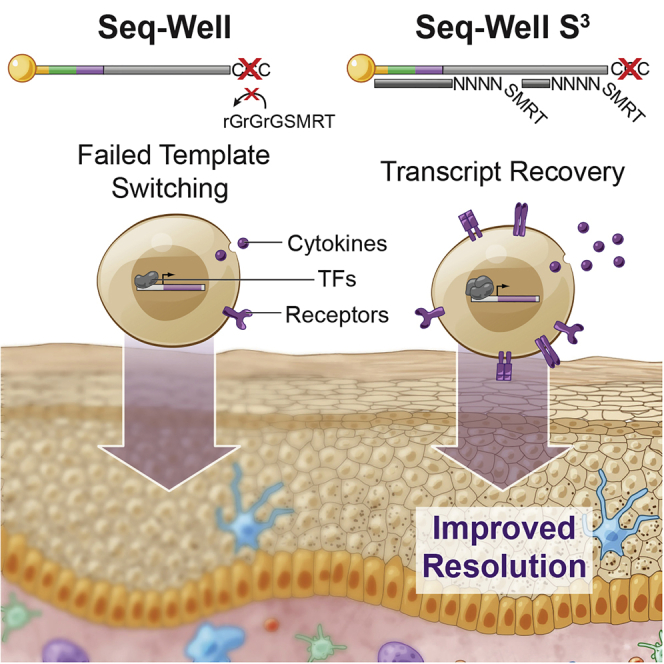
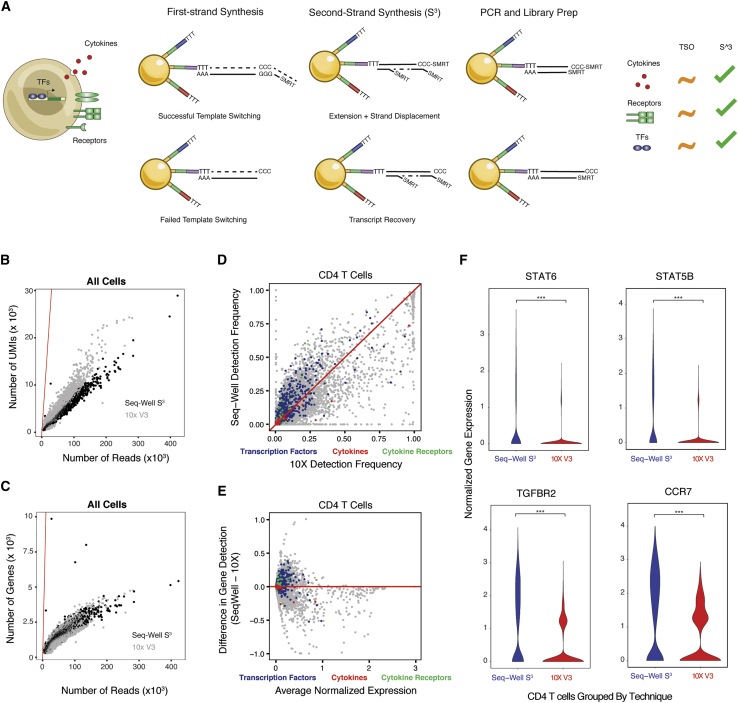
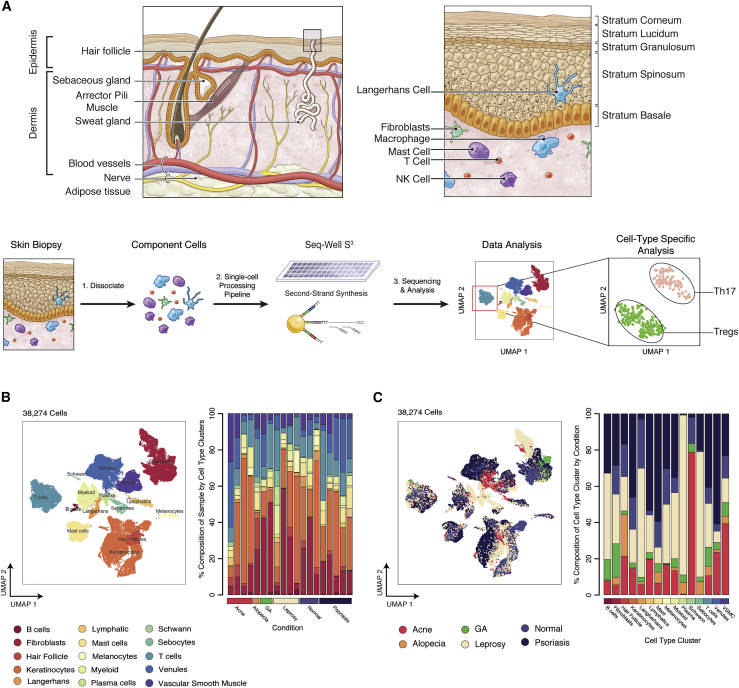
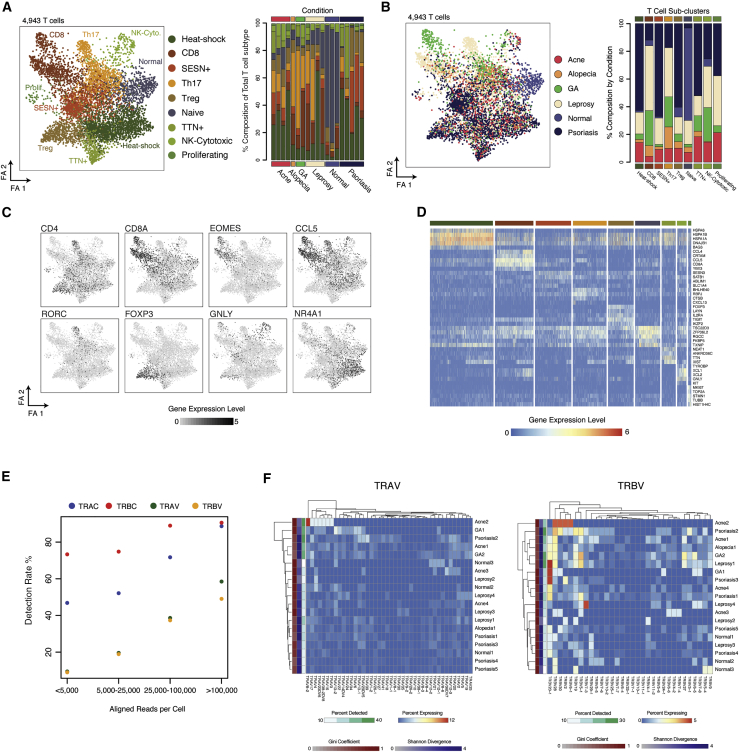
High-throughput single-cell RNA-sequencing (scRNA-seq) methodologies enable characterization of complex biological samples by increasing the number of cells that can be profiled contemporaneously. Nevertheless, these approaches recover less information per cell than low-throughput strategies. To accurately report the expression of key phenotypic features of cells, scRNA-seq platforms are needed that are both high fidelity and high throughput. To address this need, we created Seq-Well S3 ("Second-Strand Synthesis"), a massively parallel scRNA-seq protocol that uses a randomly primed second-strand synthesis to recover complementary DNA (cDNA) molecules that were successfully reverse transcribed but to which a second oligonucleotide handle, necessary for subsequent whole transcriptome amplification, was not appended due to inefficient template switching. Seq-Well S3 increased the efficiency of transcript capture and gene detection compared with that of previous iterations by up to 10- and 5-fold, respectively. We used Seq-Well S3 to chart the transcriptional landscape of five human inflammatory skin diseases, thus providing a resource for the further study of human skin inflammation.
Keywords: Seq-Well; acne; alopecia areata; granuloma annulare; leprosy; psoriasis; scRNA-seq; single-cell RNA sequencing; skin inflammation.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests A.K.S. and J.C.L. have received compensation for consulting and SAB membership from Honeycomb Biotechnologies. A.K.S. has received compensation for consulting and SAB membership from Cellarity, Repertoire Immune Medicines, Orche Bio, and Dahlia Biosciences. T.M.G., T.K.H., M.H.W., A.K.S., and J.C.L. are co-inventors on a provisional patent application filed by MIT relating to the improved methodology described in this manuscript.
Figures
References
-
- Best J.A., Blair D.A., Knell J., Yang E., Mayya V., Doedens A., Dustin M.L., Goldrath A.W., Monach P., Shinton S.A., Immunological Genome Project Consortium Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 2013;14:404–412. - PMC - PubMed
-
- Beylot C., Auffret N., Poli F., Claudel J.P., Leccia M.T., Del Giudice P., Dreno B. Propionibacterium acnes: an update on its role in the pathogenesis of acne. J. Eur. Acad. Dermatol. Venereol. 2014;28:271–278. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- F30 AI143160/AI/NIAID NIH HHS/United States
- P30 AR075043/AR/NIAMS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P30 CA014051/CA/NCI NIH HHS/United States
- R01 AR073252/AR/NIAMS NIH HHS/United States
- P01 AI039671/AI/NIAID NIH HHS/United States
- U19 AI089992/AI/NIAID NIH HHS/United States
- U2C CA233195/CA/NCI NIH HHS/United States
- R01 AI022553/AI/NIAID NIH HHS/United States
- R01 AR074302/AR/NIAMS NIH HHS/United States
- K08 AR060802/AR/NIAMS NIH HHS/United States
- N01 AI030025/AI/NIAID NIH HHS/United States
- R01 AI130025/AI/NIAID NIH HHS/United States
- P01 AI045757/AI/NIAID NIH HHS/United States
- R01 AR040312/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
