

Probabilistic Estimation of Identity by Descent Segment Endpoints and Detection of Recent Selection
- PMID: 33053335
- PMCID: PMC7553009
- DOI: 10.1016/j.ajhg.2020.09.010
Probabilistic Estimation of Identity by Descent Segment Endpoints and Detection of Recent Selection
Abstract
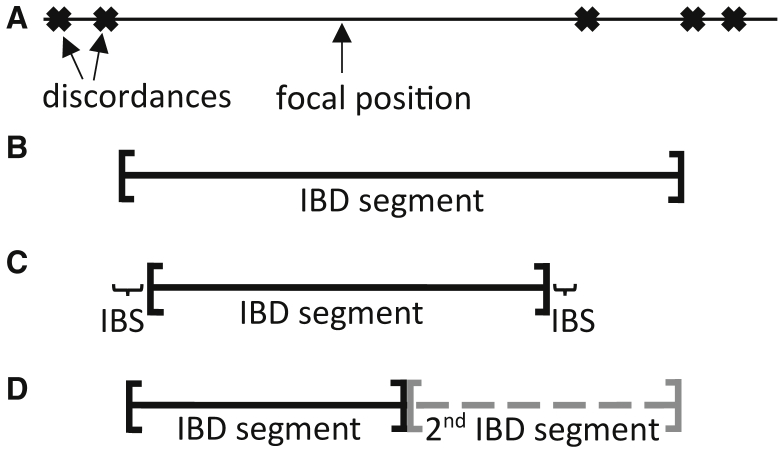
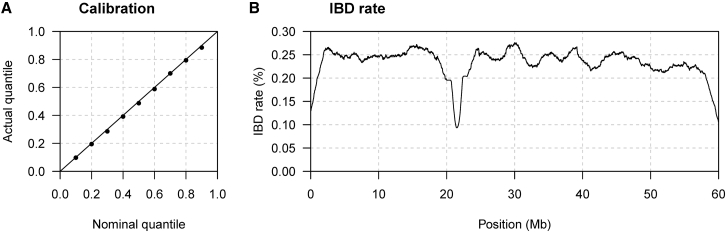
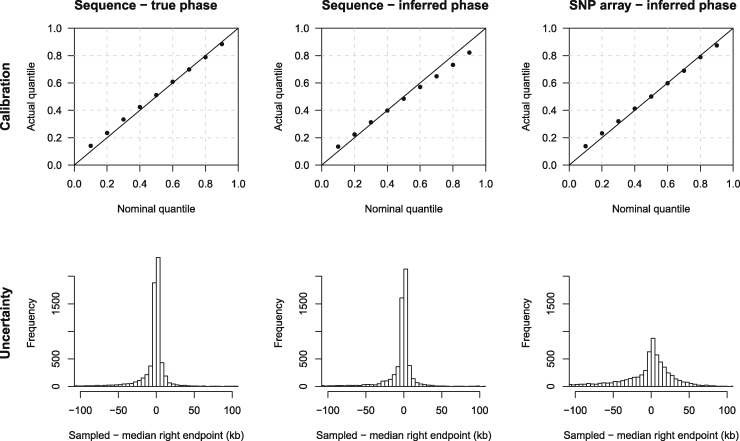
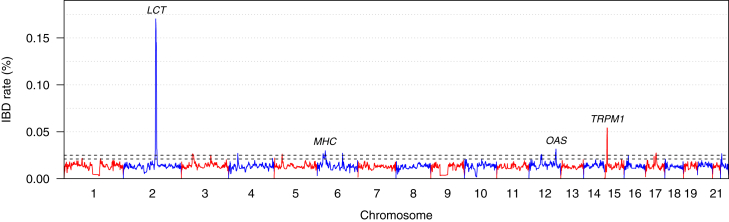
Most methods for fast detection of identity by descent (IBD) segments report identity by state segments without any quantification of the uncertainty in the endpoints and lengths of the IBD segments. We present a method for determining the posterior probability distribution of IBD segment endpoints. Our approach accounts for genotype errors, recent mutations, and gene conversions which disrupt DNA sequence identity within IBD segments, and it can be applied to large cohorts with whole-genome sequence or SNP array data. We find that our method's estimates of uncertainty are well calibrated for homogeneous samples. We quantify endpoint uncertainty for 77.7 billion IBD segments from 408,883 individuals of white British ancestry in the UK Biobank, and we use these IBD segments to find regions showing evidence of recent natural selection. We show that many spurious selection signals are eliminated by the use of unbiased estimates of IBD segment endpoints and a pedigree-based genetic map. Eleven of the twelve regions with the greatest evidence for recent selection in our scan have been identified as selected in previous analyses using different approaches. Our computationally efficient method for quantifying IBD segment endpoint uncertainty is implemented in the open source ibd-ends software package.
Keywords: identity by descent; natural selection; recent positive selection.
Copyright © 2020 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
