

A porcine model of phenylketonuria generated by CRISPR/Cas9 genome editing
- PMID: 33055427
- PMCID: PMC7605535
- DOI: 10.1172/jci.insight.141523
A porcine model of phenylketonuria generated by CRISPR/Cas9 genome editing
Abstract
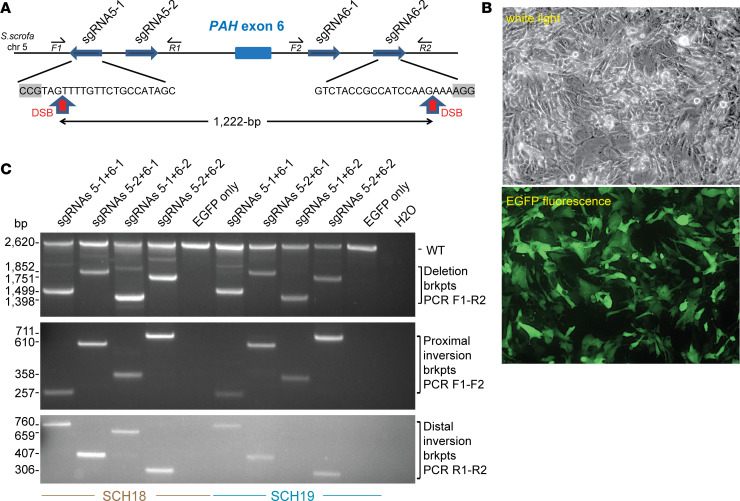
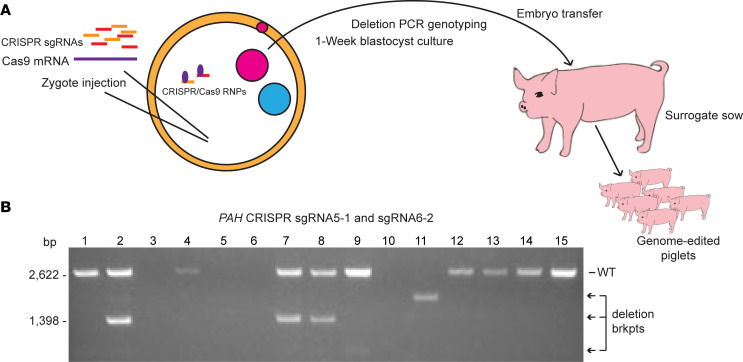
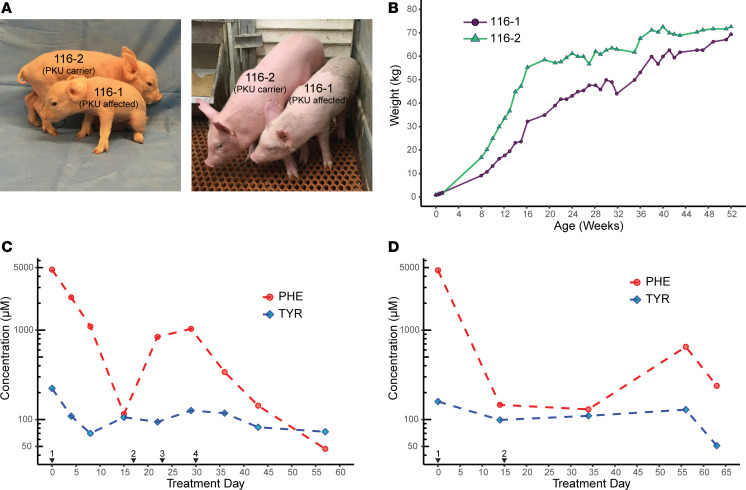
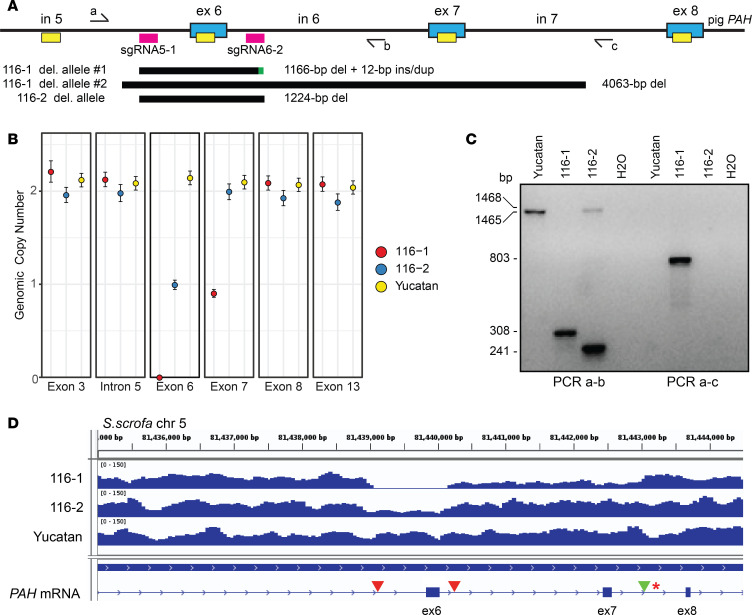
Phenylalanine hydroxylase-deficient (PAH-deficient) phenylketonuria (PKU) results in systemic hyperphenylalaninemia, leading to neurotoxicity with severe developmental disabilities. Dietary phenylalanine (Phe) restriction prevents the most deleterious effects of hyperphenylalaninemia, but adherence to diet is poor in adult and adolescent patients, resulting in characteristic neurobehavioral phenotypes. Thus, an urgent need exists for new treatments. Additionally, rodent models of PKU do not adequately reflect neurocognitive phenotypes, and thus there is a need for improved animal models. To this end, we have developed PAH-null pigs. After selection of optimal CRISPR/Cas9 genome-editing reagents by using an in vitro cell model, zygote injection of 2 sgRNAs and Cas9 mRNA demonstrated deletions in preimplantation embryos, with embryo transfer to a surrogate leading to 2 founder animals. One pig was heterozygous for a PAH exon 6 deletion allele, while the other was compound heterozygous for deletions of exon 6 and of exons 6-7. The affected pig exhibited hyperphenylalaninemia (2000-5000 μM) that was treatable by dietary Phe restriction, consistent with classical PKU, along with juvenile growth retardation, hypopigmentation, ventriculomegaly, and decreased brain gray matter volume. In conclusion, we have established a large-animal preclinical model of PKU to investigate pathophysiology and to assess new therapeutic interventions.
Keywords: Amino acid metabolism; Genetic diseases; Genetics; Metabolism; Mouse models.
Conflict of interest statement
Figures

References
-
- Donlon J, Sarkissian C, Levy H, Scriver CR. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Valle D, Antonarakis S, Ballabio A, Beaudet A, Mitchell GA, eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; Accessed August 10, 2020. https://ommbid-mhmedical-com.pitt.idm.oclc.org/content.aspx?bookid=2709&....
-
- Scriver C, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Beaudet A, et al. eds. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; 2001:1667–1724.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
