

Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells
- PMID: 33057200
- PMCID: PMC7606745
- DOI: 10.1038/s41586-020-2786-7
Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells
Abstract
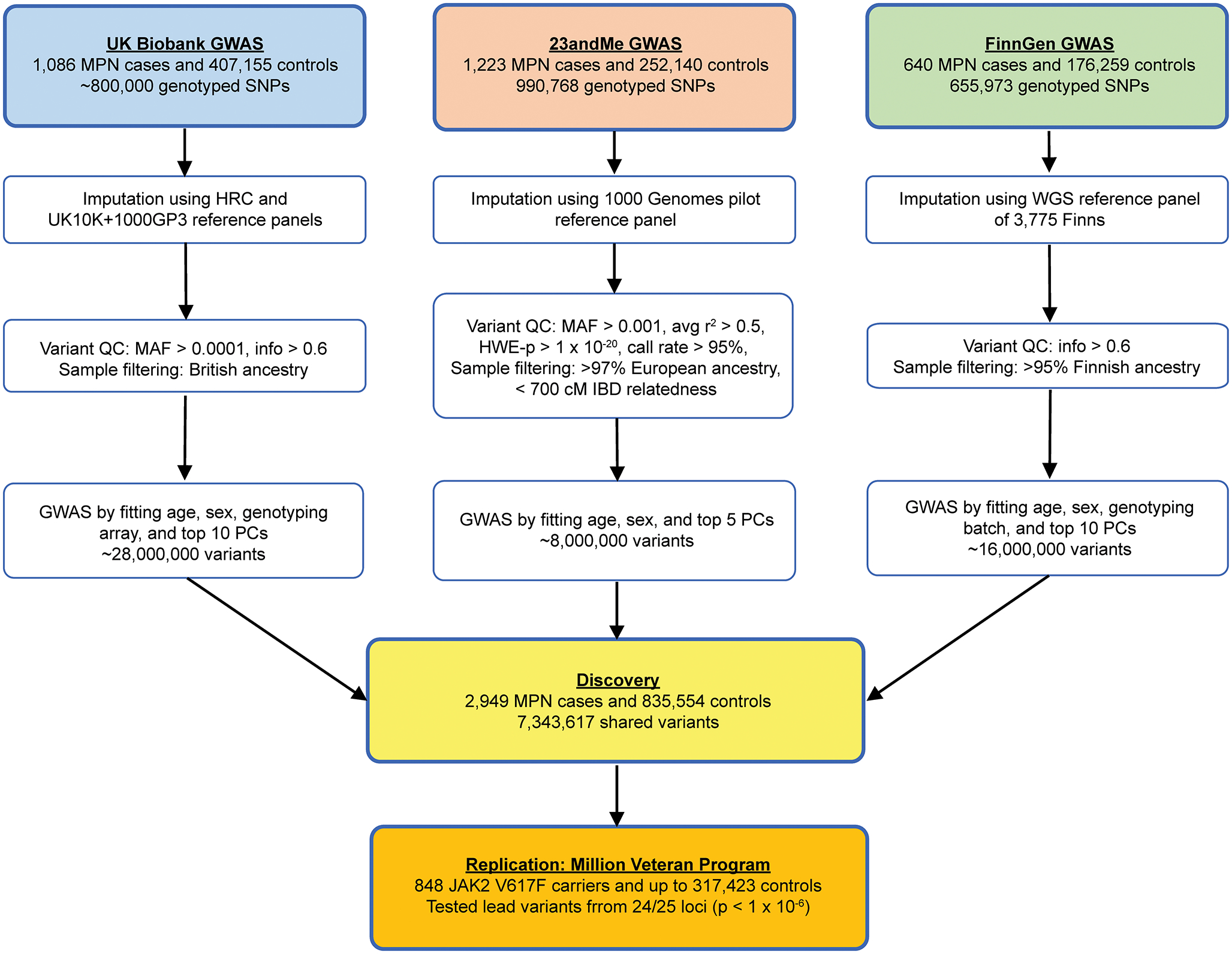
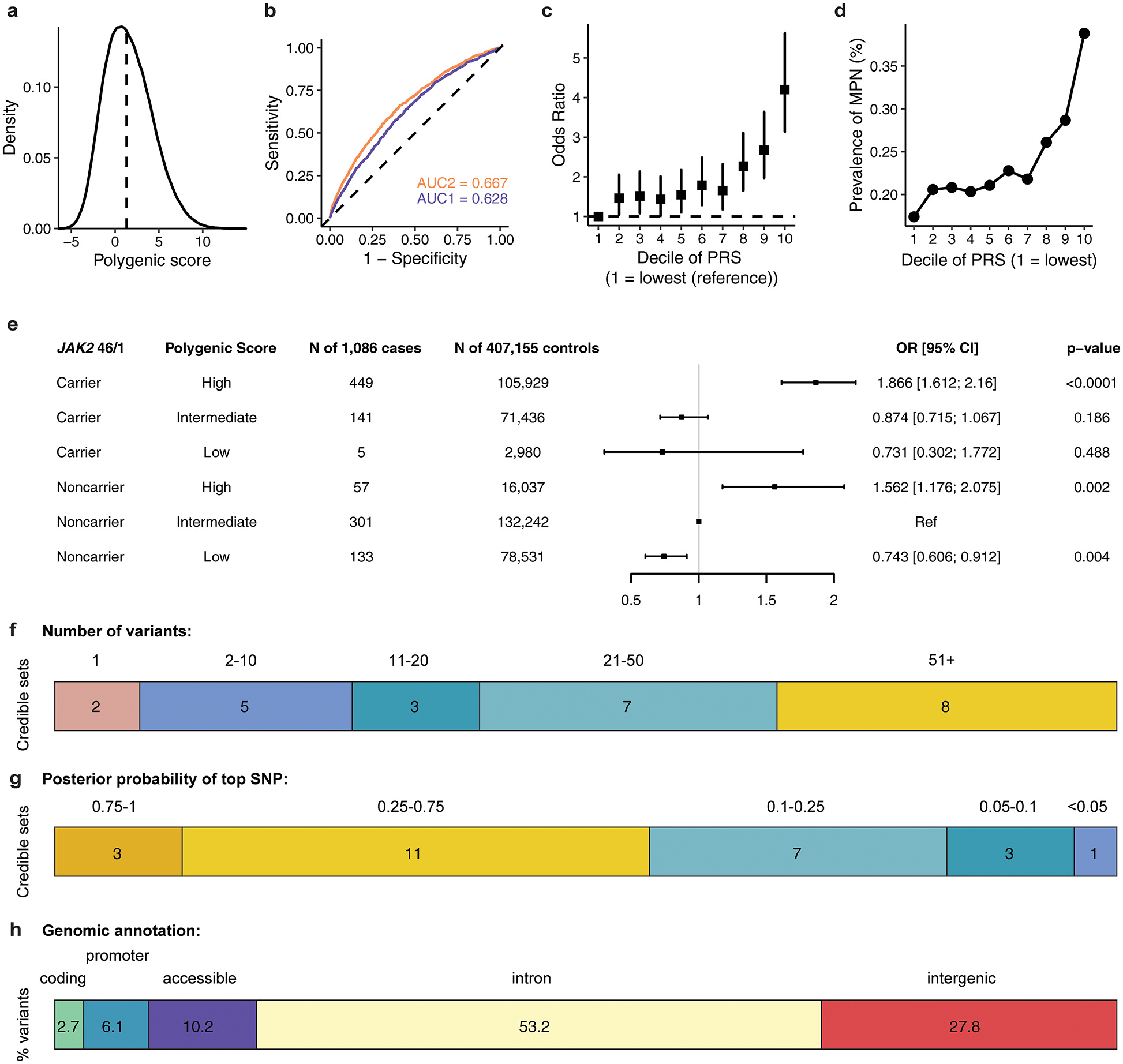
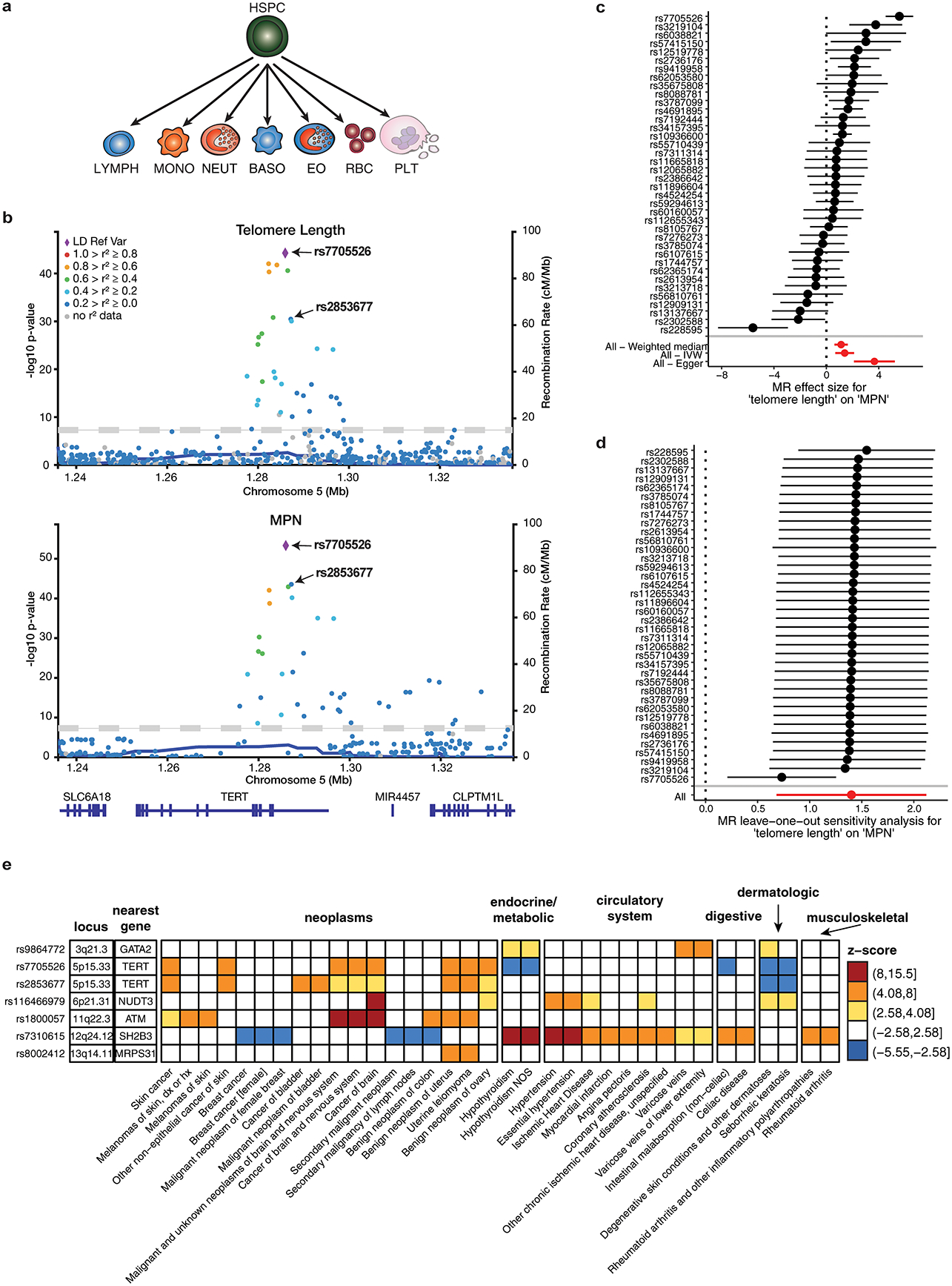
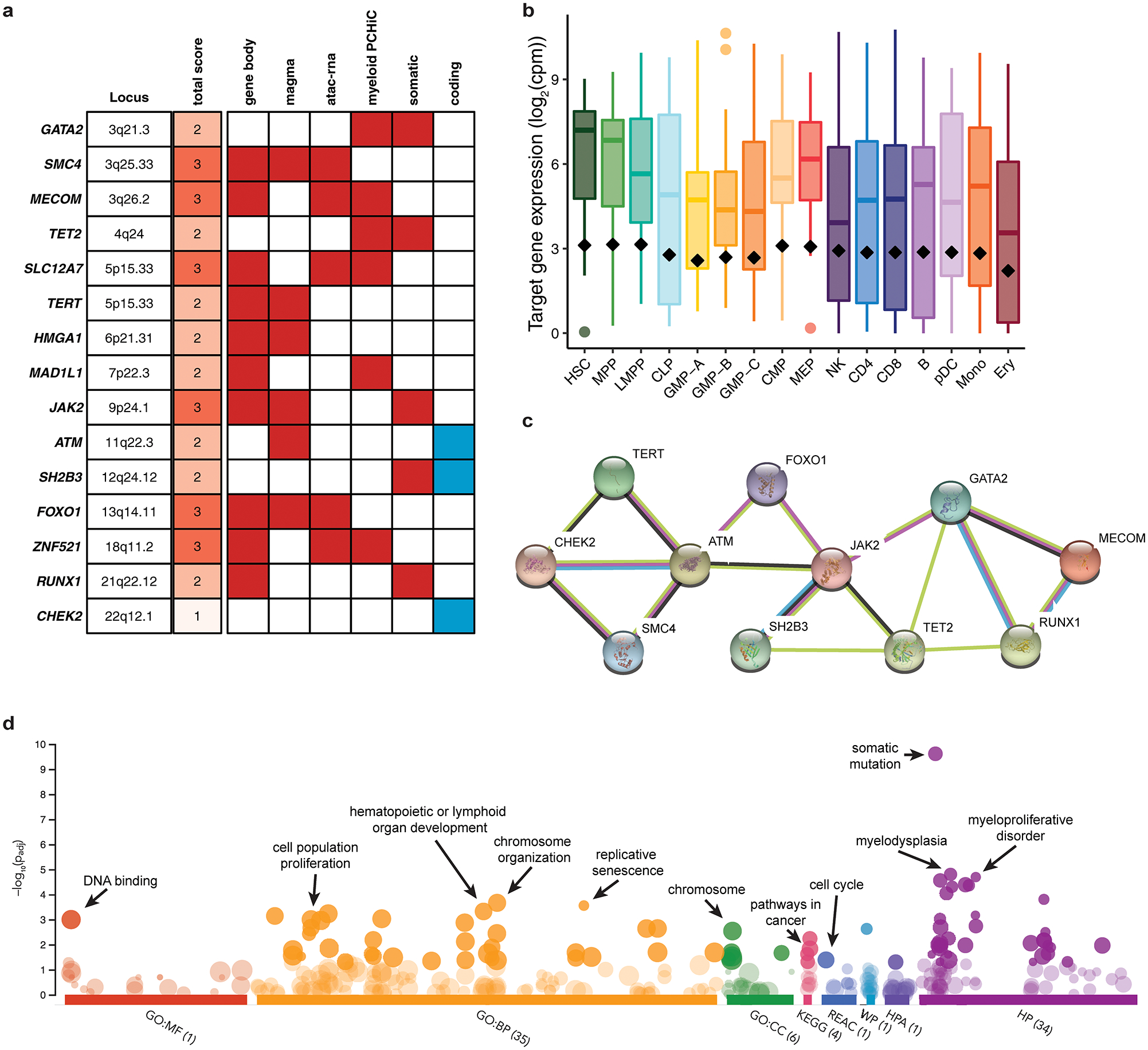
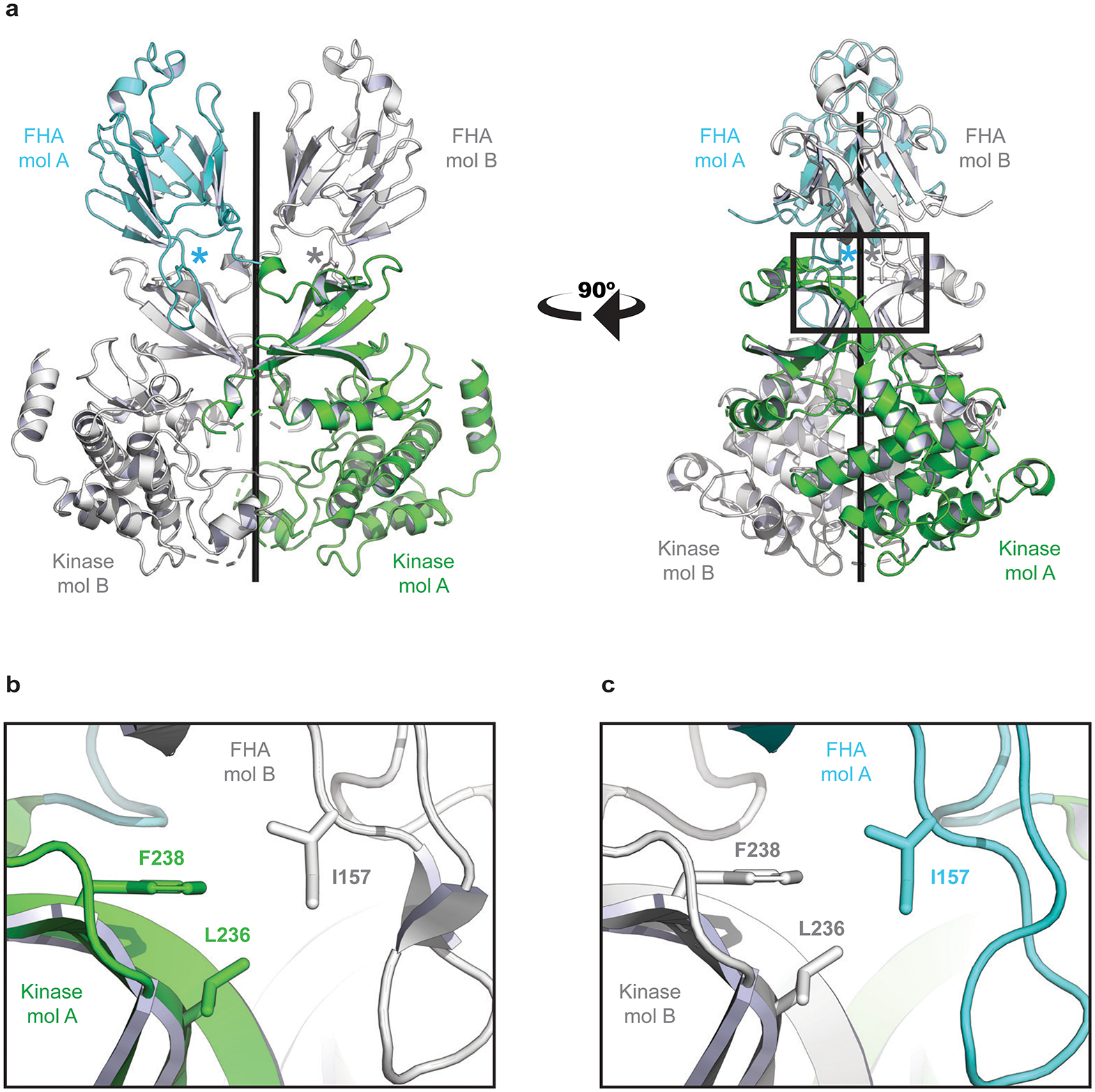
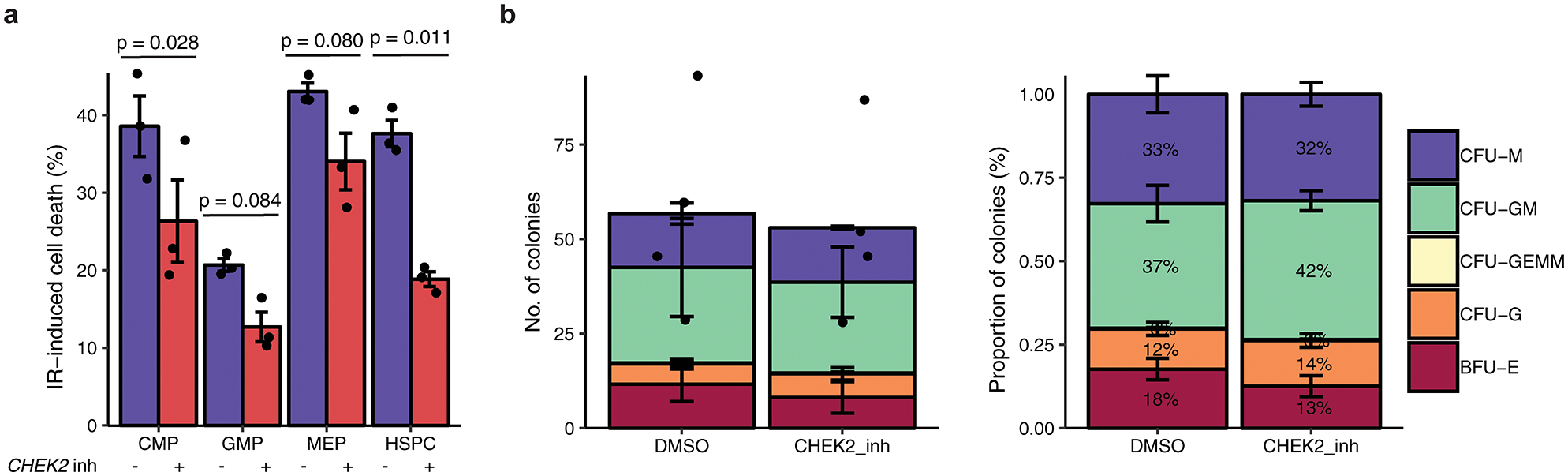
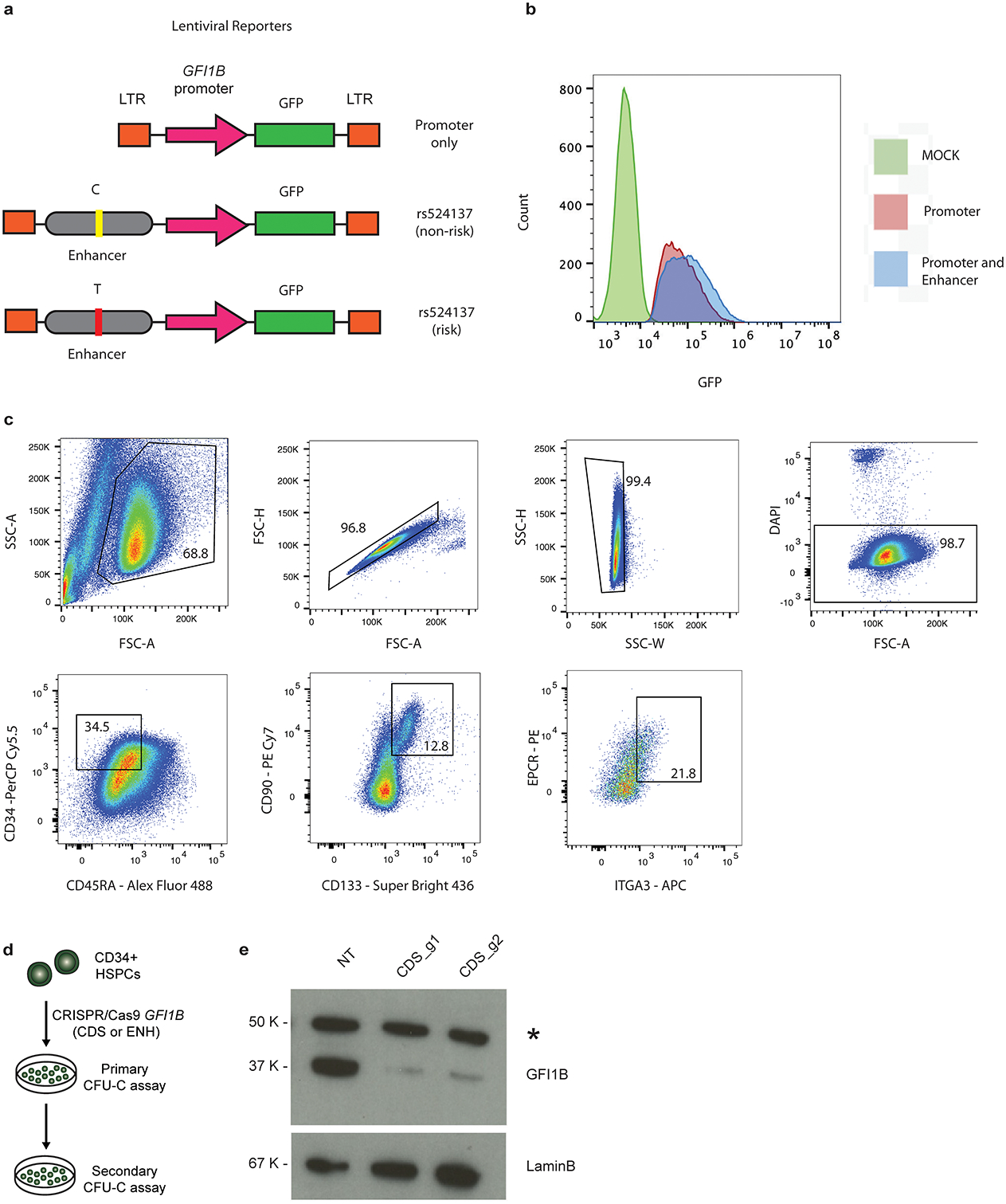
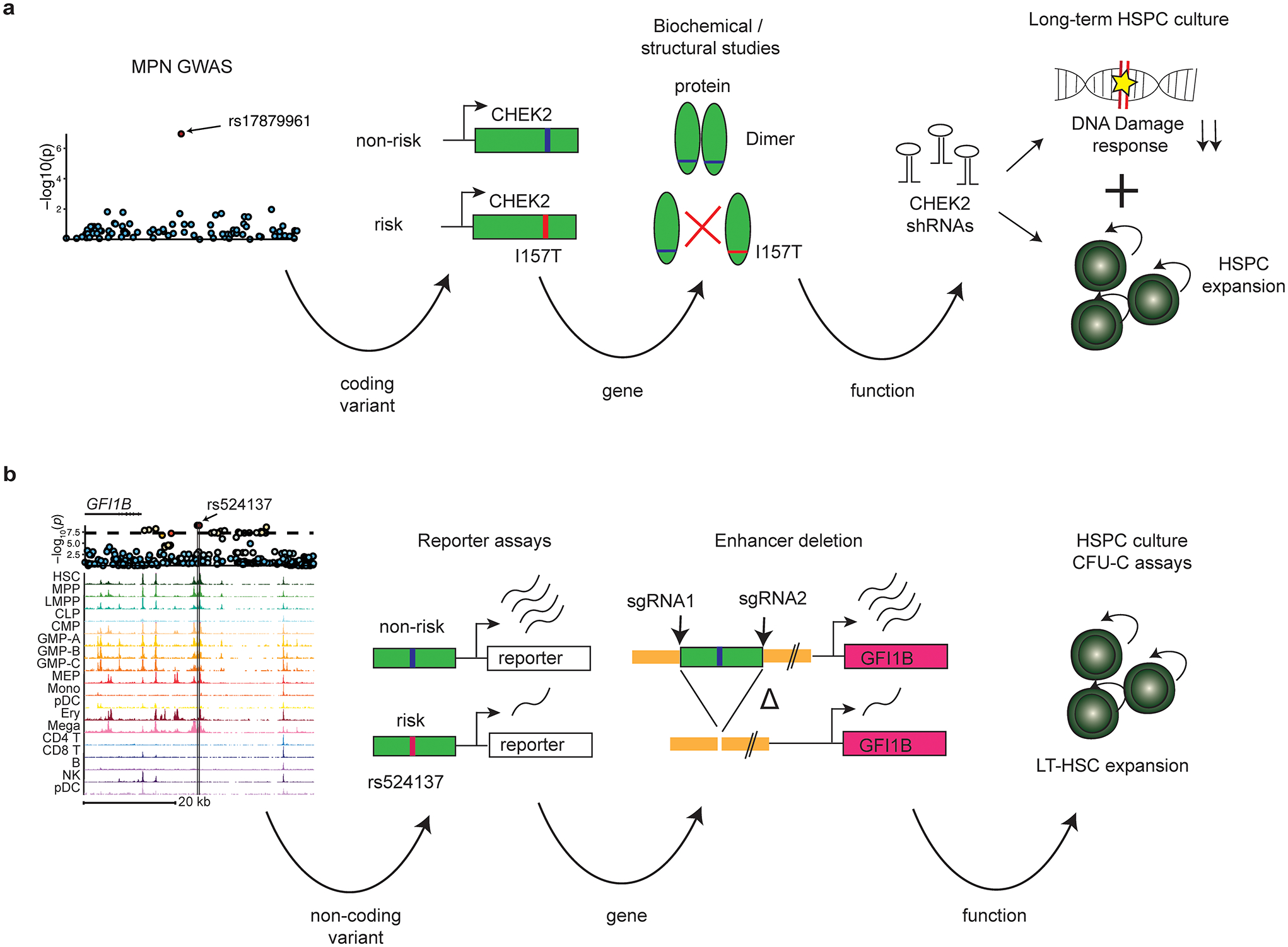
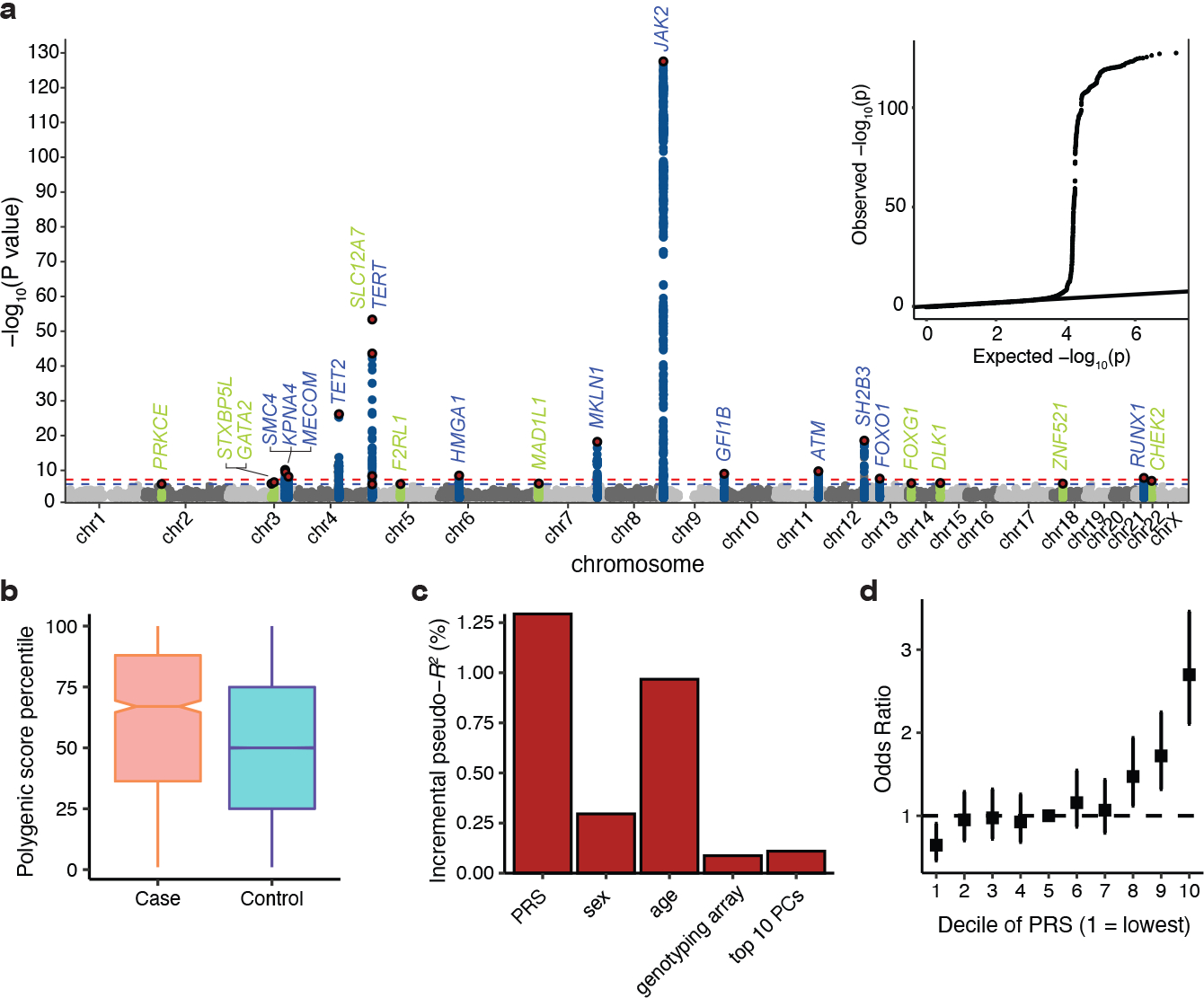
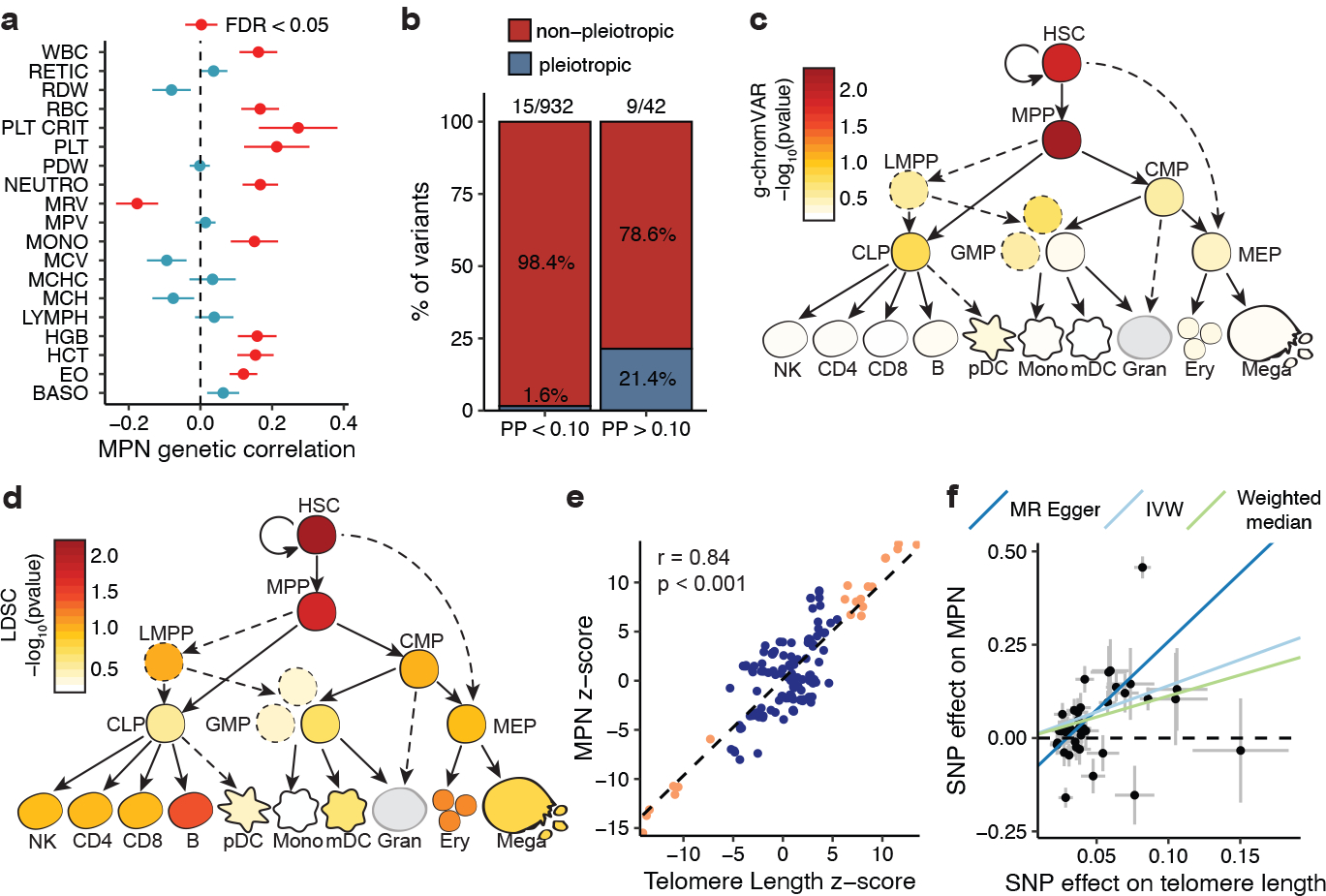
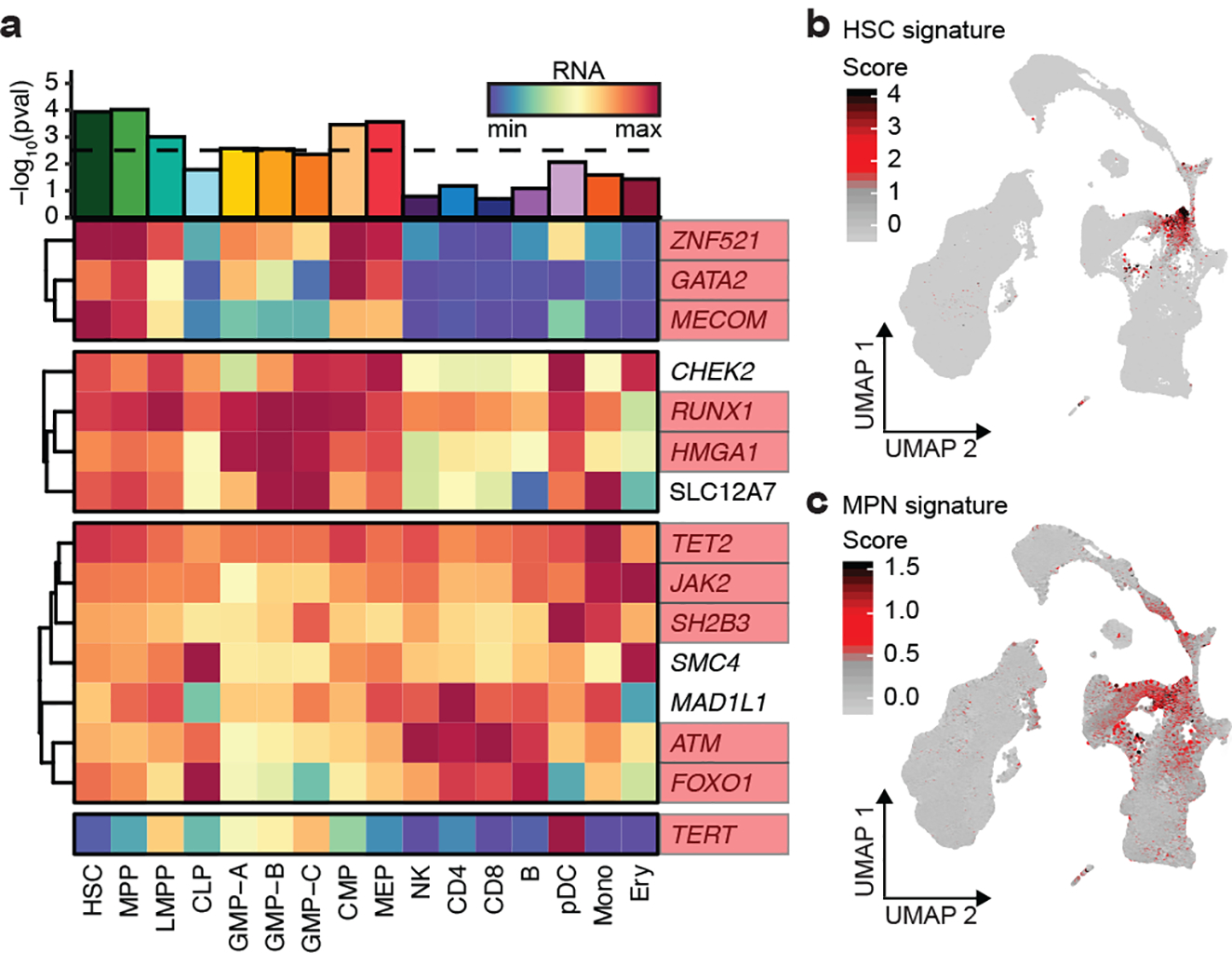
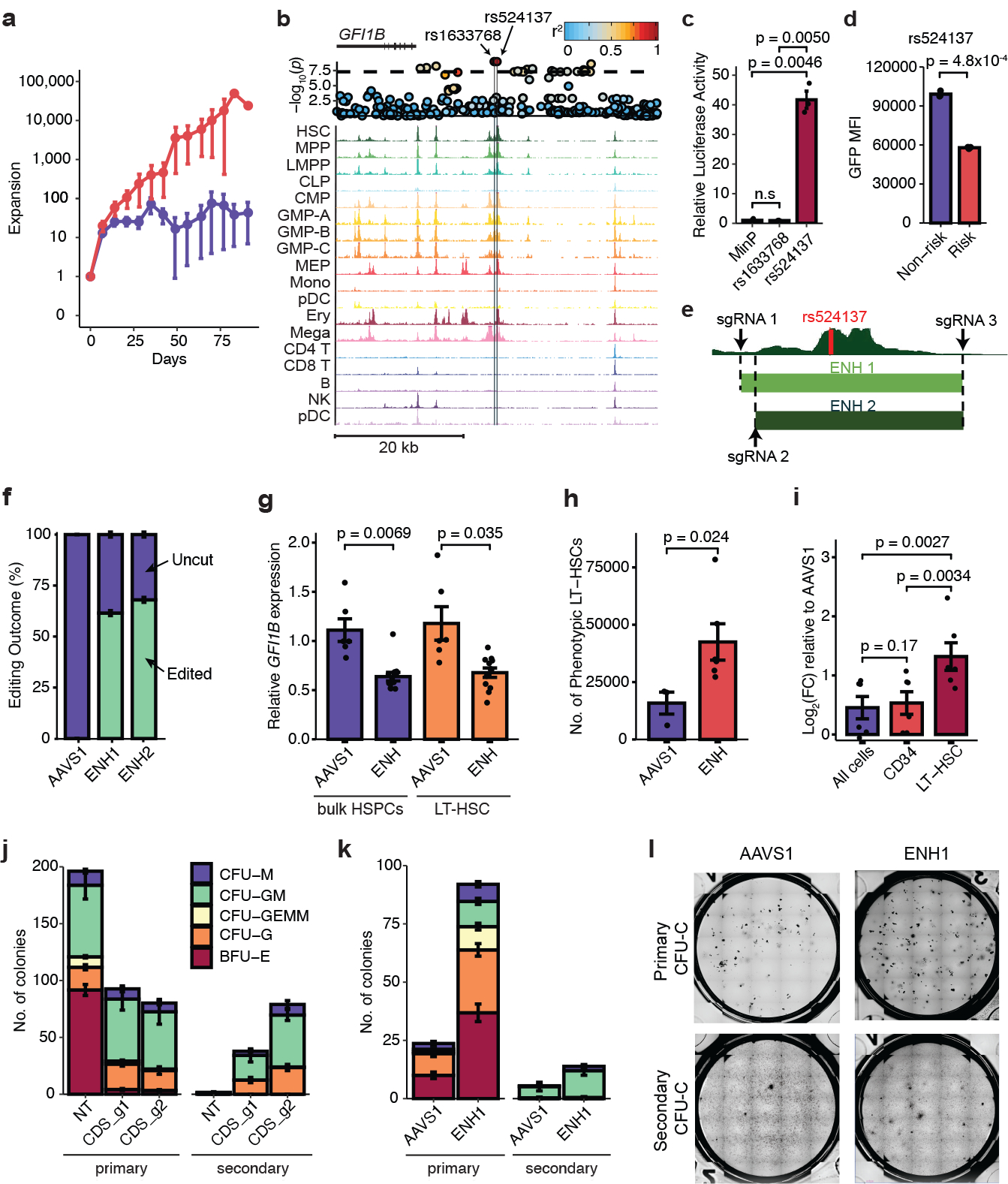
Myeloproliferative neoplasms (MPNs) are blood cancers that are characterized by the excessive production of mature myeloid cells and arise from the acquisition of somatic driver mutations in haematopoietic stem cells (HSCs). Epidemiological studies indicate a substantial heritable component of MPNs that is among the highest known for cancers1. However, only a limited number of genetic risk loci have been identified, and the underlying biological mechanisms that lead to the acquisition of MPNs remain unclear. Here, by conducting a large-scale genome-wide association study (3,797 cases and 1,152,977 controls), we identify 17 MPN risk loci (P < 5.0 × 10-8), 7 of which have not been previously reported. We find that there is a shared genetic architecture between MPN risk and several haematopoietic traits from distinct lineages; that there is an enrichment for MPN risk variants within accessible chromatin of HSCs; and that increased MPN risk is associated with longer telomere length in leukocytes and other clonal haematopoietic states-collectively suggesting that MPN risk is associated with the function and self-renewal of HSCs. We use gene mapping to identify modulators of HSC biology linked to MPN risk, and show through targeted variant-to-function assays that CHEK2 and GFI1B have roles in altering the function of HSCs to confer disease risk. Overall, our results reveal a previously unappreciated mechanism for inherited MPN risk through the modulation of HSC function.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
