

IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring
- PMID: 33057676
- PMCID: PMC7566497
- DOI: 10.1093/gigascience/giaa111
IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring
Abstract
Background: Metagenomic next-generation sequencing (mNGS) has enabled the rapid, unbiased detection and identification of microbes without pathogen-specific reagents, culturing, or a priori knowledge of the microbial landscape. mNGS data analysis requires a series of computationally intensive processing steps to accurately determine the microbial composition of a sample. Existing mNGS data analysis tools typically require bioinformatics expertise and access to local server-class hardware resources. For many research laboratories, this presents an obstacle, especially in resource-limited environments.
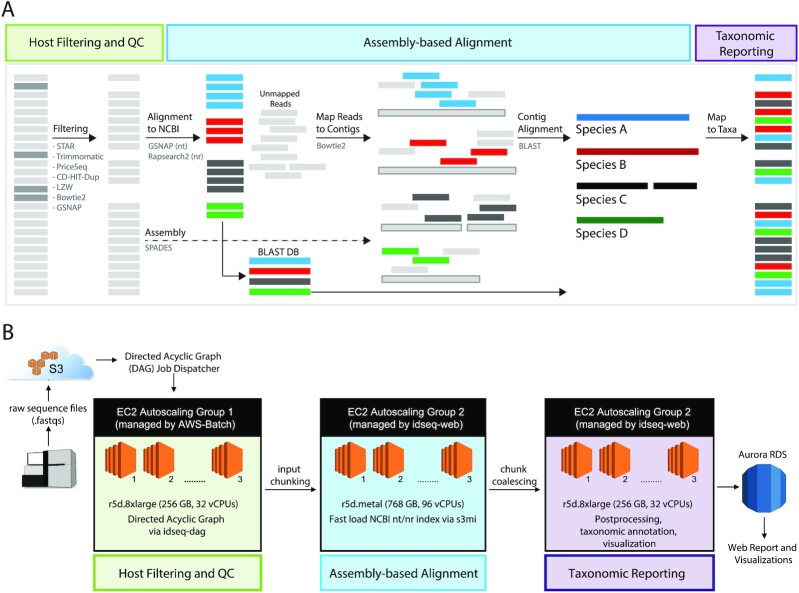
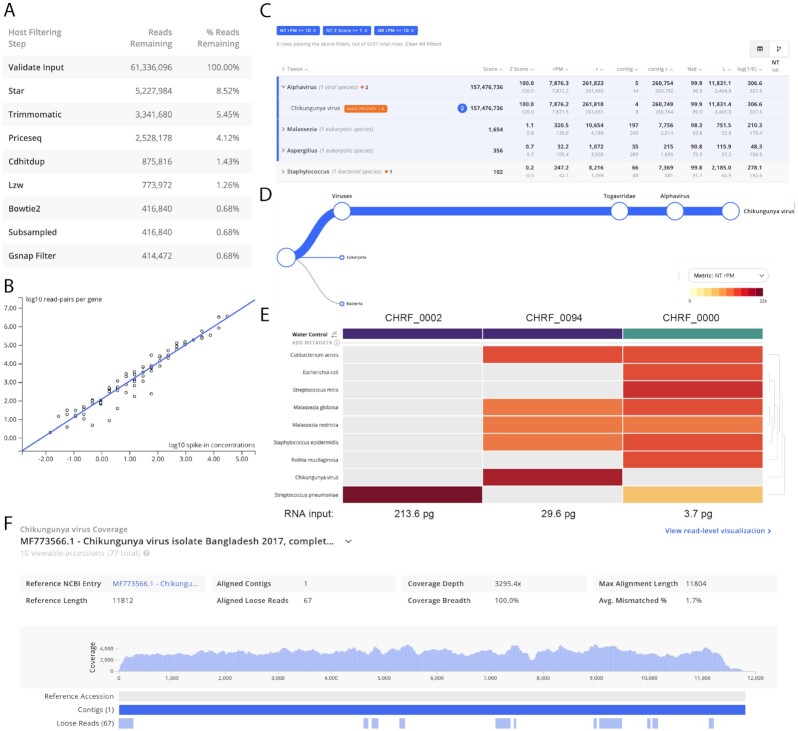
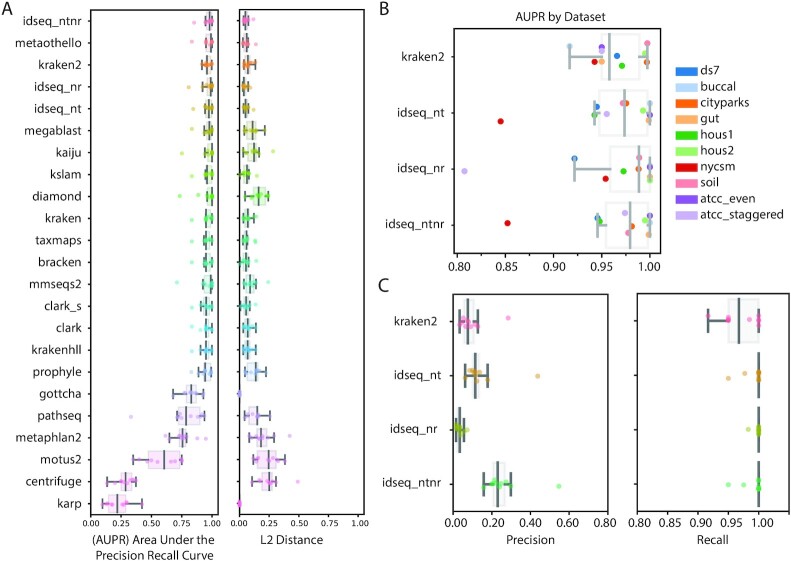
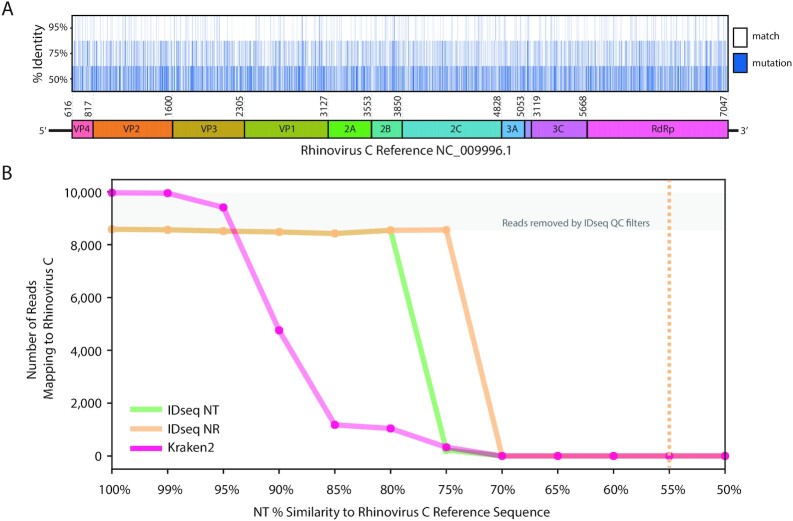
Findings: We present IDseq, an open source cloud-based metagenomics pipeline and service for global pathogen detection and monitoring (https://idseq.net). The IDseq Portal accepts raw mNGS data, performs host and quality filtration steps, then executes an assembly-based alignment pipeline, which results in the assignment of reads and contigs to taxonomic categories. The taxonomic relative abundances are reported and visualized in an easy-to-use web application to facilitate data interpretation and hypothesis generation. Furthermore, IDseq supports environmental background model generation and automatic internal spike-in control recognition, providing statistics that are critical for data interpretation. IDseq was designed with the specific intent of detecting novel pathogens. Here, we benchmark novel virus detection capability using both synthetically evolved viral sequences and real-world samples, including IDseq analysis of a nasopharyngeal swab sample acquired and processed locally in Cambodia from a tourist from Wuhan, China, infected with the recently emergent SARS-CoV-2.
Conclusion: The IDseq Portal reduces the barrier to entry for mNGS data analysis and enables bench scientists, clinicians, and bioinformaticians to gain insight from mNGS datasets for both known and novel pathogens.
Keywords: COVID-2019; cloud-based; metagenomics; pathogen detection; virus.
© The Author(s) 2020. Published by Oxford University Press GigaScience.
Figures
References
-
- Lu J, Breitwieser FP, Thielen P, et al. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput Sci. 2017;3:e104.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
