

In silico benchmarking of metagenomic tools for coding sequence detection reveals the limits of sensitivity and precision
- PMID: 33059593
- PMCID: PMC7559173
- DOI: 10.1186/s12859-020-03802-0
In silico benchmarking of metagenomic tools for coding sequence detection reveals the limits of sensitivity and precision
Abstract
Background: High-throughput sequencing can establish the functional capacity of a microbial community by cataloging the protein-coding sequences (CDS) present in the metagenome of the community. The relative performance of different computational methods for identifying CDS from whole-genome shotgun sequencing is not fully established.
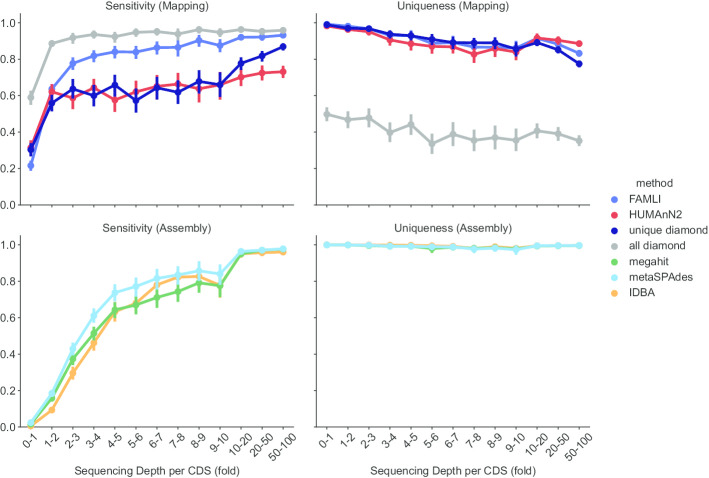
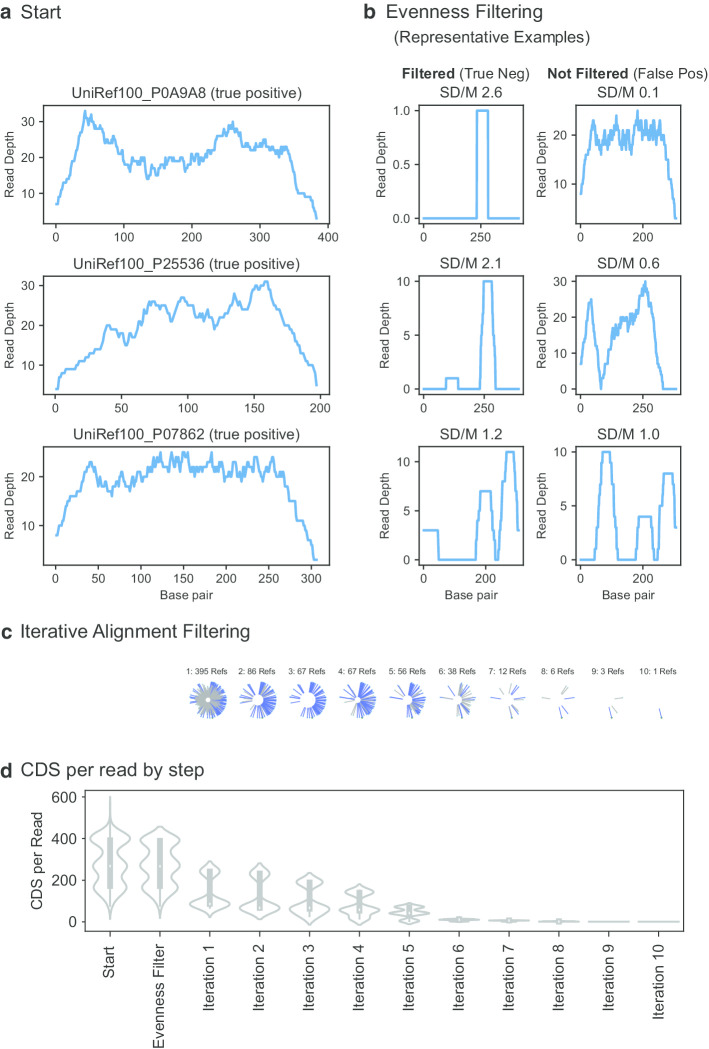
Results: Here we present an automated benchmarking workflow, using synthetic shotgun sequencing reads for which we know the true CDS content of the underlying communities, to determine the relative performance (sensitivity, positive predictive value or PPV, and computational efficiency) of different metagenome analysis tools for extracting the CDS content of a microbial community. Assembly-based methods are limited by coverage depth, with poor sensitivity for CDS at < 5X depth of sequencing, but have excellent PPV. Mapping-based techniques are more sensitive at low coverage depths, but can struggle with PPV. We additionally describe an expectation maximization based iterative algorithmic approach which we show to successfully improve the PPV of a mapping based technique while retaining improved sensitivity and computational efficiency.
Conclusion: Our benchmarking approach reveals the trade-offs of assembly versus alignment-based approaches and the relative performance of specific implementations when one wishes to extract the protein coding capacity of microbial communities.
Keywords: Bioinformatics; Metagenomics; Microbiome.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures

References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
