

PRIM1 deficiency causes a distinctive primordial dwarfism syndrome
- PMID: 33060134
- PMCID: PMC7608753
- DOI: 10.1101/gad.340190.120
PRIM1 deficiency causes a distinctive primordial dwarfism syndrome
Abstract
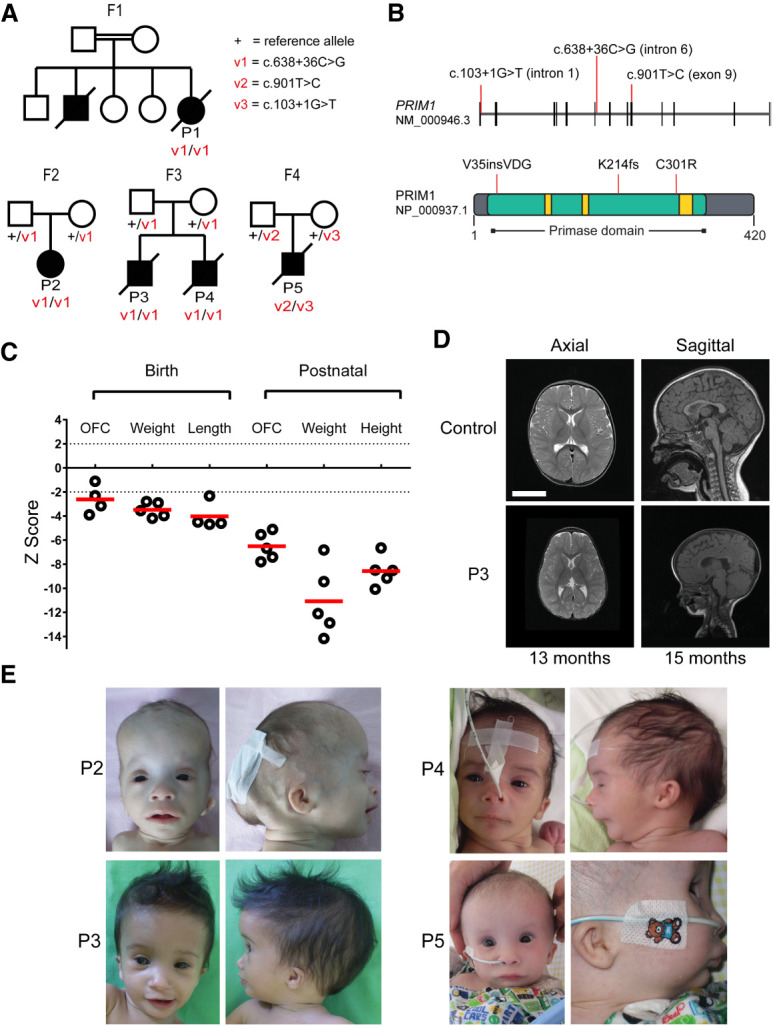
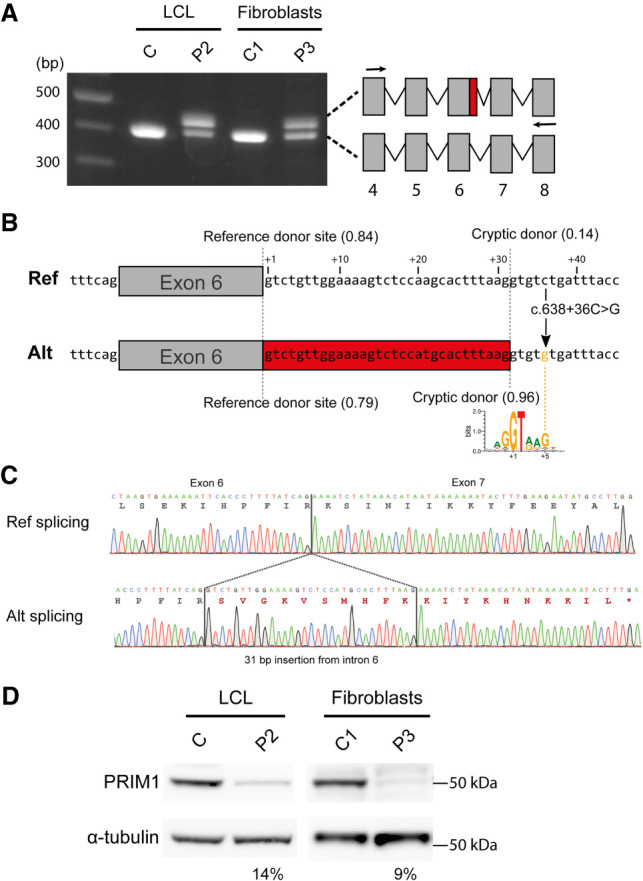
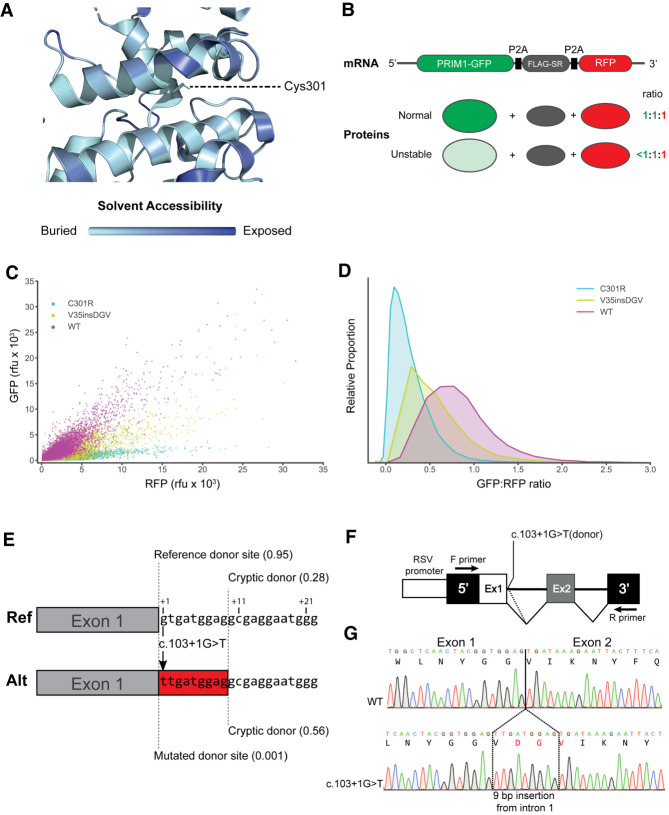
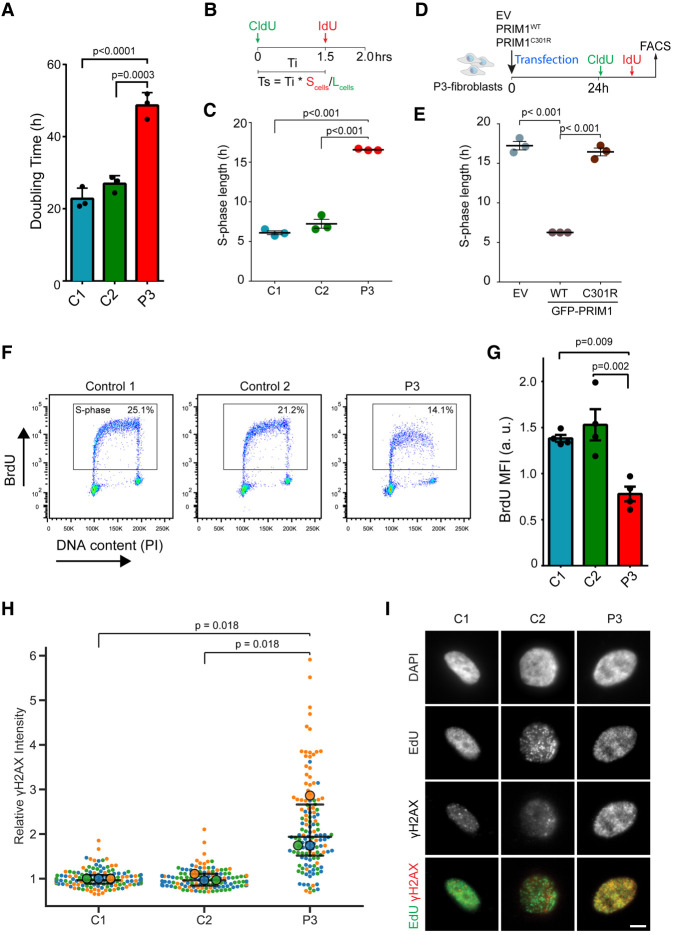
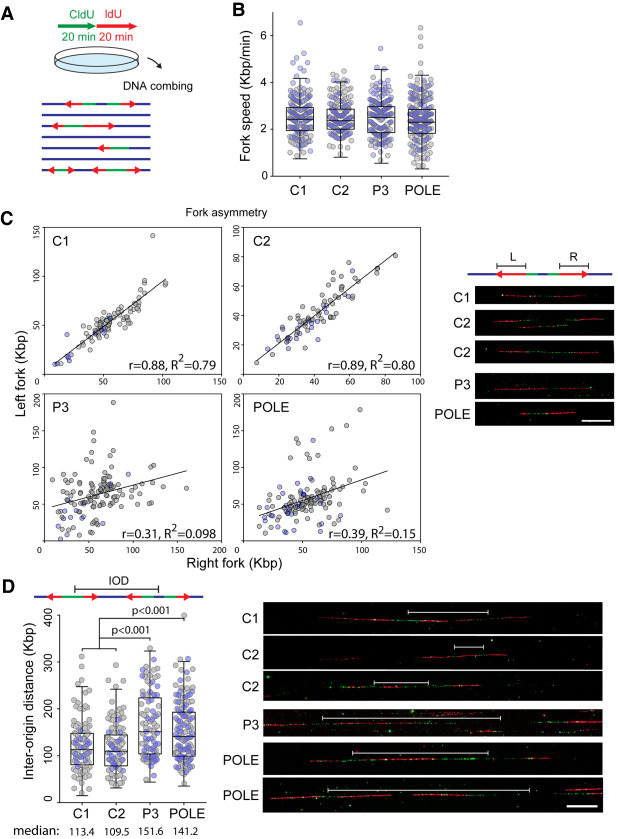
DNA replication is fundamental for cell proliferation in all organisms. Nonetheless, components of the replisome have been implicated in human disease, and here we report PRIM1 encoding the catalytic subunit of DNA primase as a novel disease gene. Using a variant classification agnostic approach, biallelic mutations in PRIM1 were identified in five individuals. PRIM1 protein levels were markedly reduced in patient cells, accompanied by replication fork asymmetry, increased interorigin distances, replication stress, and prolonged S-phase duration. Consequently, cell proliferation was markedly impaired, explaining the patients' extreme growth failure. Notably, phenotypic features distinct from those previously reported with DNA polymerase genes were evident, highlighting differing developmental requirements for this core replisome component that warrant future investigation.
Keywords: DNA replication; genome stability; growth disorders; human genetics; rare disease.
© 2020 Parry et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bicknell LS, Walker S, Klingseisen A, Stiff T, Leitch A, Kerzendorfer C, Martin C-A, Yeyati P, Al Sanna N, Bober M, et al. 2011b. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet 43: 350–355. 10.1038/ng.776 - DOI - PubMed
-
- Burrage LC, Charng WL, Eldomery MK, Willer JR, Davis EE, Lugtenberg D, Zhu W, Leduc MS, Akdemir ZC, Azamian M, et al. 2015. De novo GMNN mutations cause autosomal-dominant primordial dwarfism associated with Meier-Gorlin syndrome. Am J Hum Genet 97: 904–913. 10.1016/j.ajhg.2015.11.006 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials