

Selective inhibition of CDK7 reveals high-confidence targets and new models for TFIIH function in transcription
- PMID: 33060135
- PMCID: PMC7608751
- DOI: 10.1101/gad.341545.120
Selective inhibition of CDK7 reveals high-confidence targets and new models for TFIIH function in transcription
Abstract
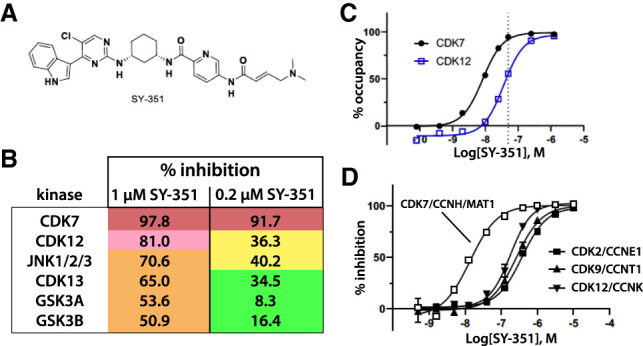
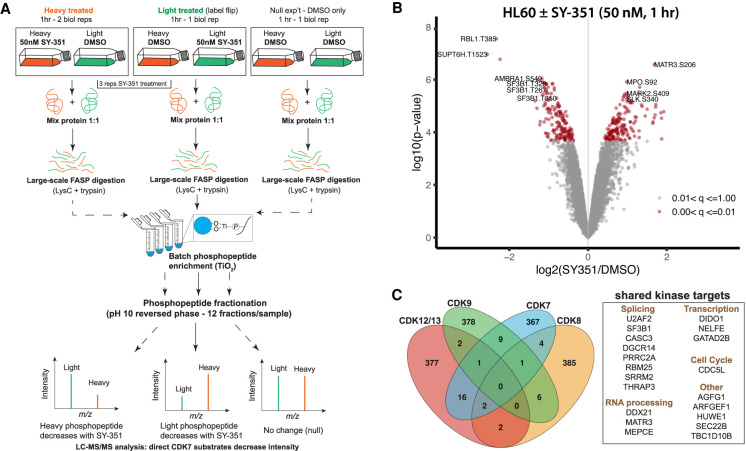
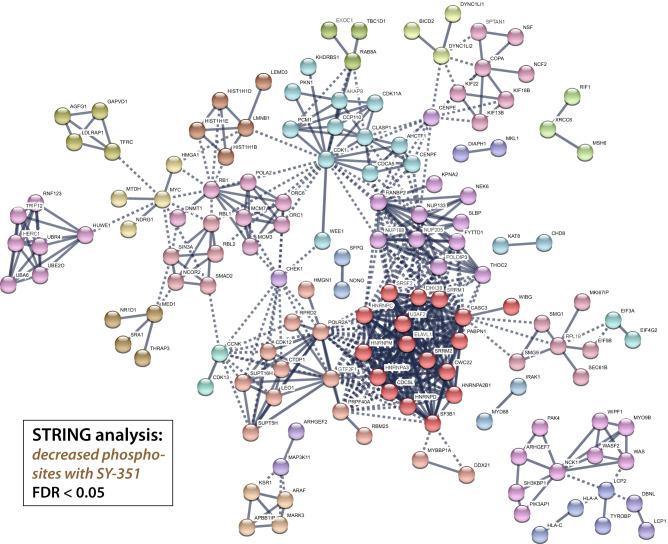
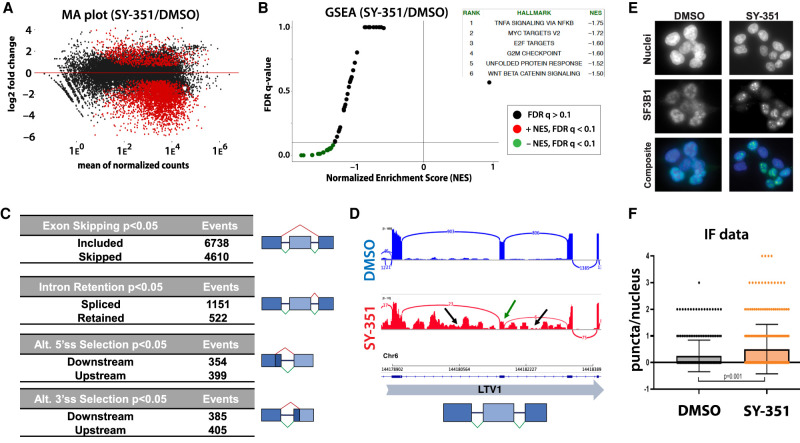
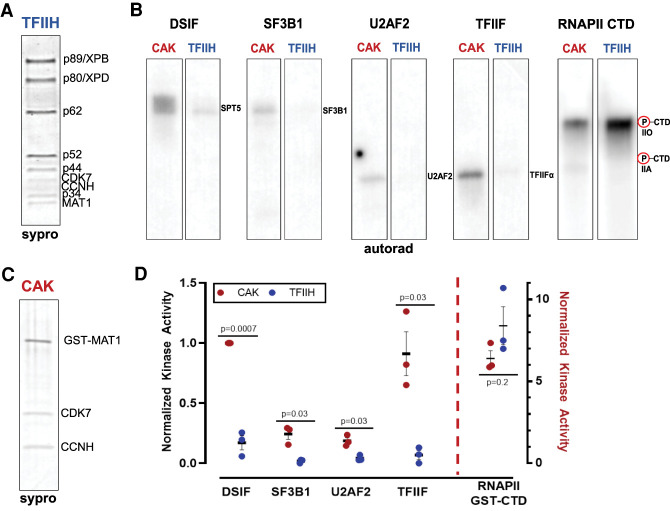
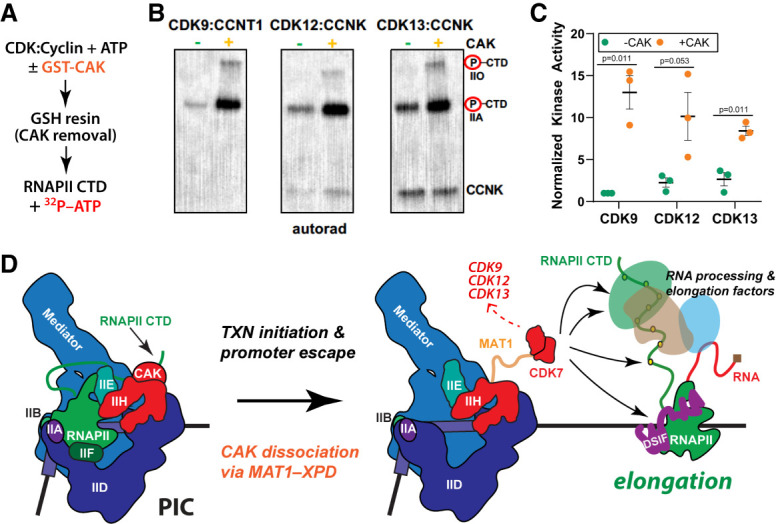
CDK7 associates with the 10-subunit TFIIH complex and regulates transcription by phosphorylating the C-terminal domain (CTD) of RNA polymerase II (RNAPII). Few additional CDK7 substrates are known. Here, using the covalent inhibitor SY-351 and quantitative phosphoproteomics, we identified CDK7 kinase substrates in human cells. Among hundreds of high-confidence targets, the vast majority are unique to CDK7 (i.e., distinct from other transcription-associated kinases), with a subset that suggest novel cellular functions. Transcription-associated factors were predominant CDK7 substrates, including SF3B1, U2AF2, and other splicing components. Accordingly, widespread and diverse splicing defects, such as alternative exon inclusion and intron retention, were characterized in CDK7-inhibited cells. Combined with biochemical assays, we establish that CDK7 directly activates other transcription-associated kinases CDK9, CDK12, and CDK13, invoking a "master regulator" role in transcription. We further demonstrate that TFIIH restricts CDK7 kinase function to the RNAPII CTD, whereas other substrates (e.g., SPT5 and SF3B1) are phosphorylated by the three-subunit CDK-activating kinase (CAK; CCNH, MAT1, and CDK7). These results suggest new models for CDK7 function in transcription and implicate CAK dissociation from TFIIH as essential for kinase activation. This straightforward regulatory strategy ensures CDK7 activation is spatially and temporally linked to transcription, and may apply toward other transcription-associated kinases.
Keywords: CDK12; CDK13; CDK7; CDK9; SF3B1; SILAC-MS; TFIIH; kinase inhibitor; splicing; transcription.
© 2020 Rimel et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous