

Neurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics
- PMID: 33060286
- PMCID: PMC8328565
- DOI: 10.1136/jmedgenet-2020-106901
Neurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics
Abstract
Background: Fetal akinesia and arthrogryposis are clinically and genetically heterogeneous and have traditionally been refractive to genetic diagnosis. The widespread availability of affordable genome-wide sequencing has facilitated accurate genetic diagnosis and gene discovery in these conditions.
Methods: We performed next generation sequencing (NGS) in 190 probands with a diagnosis of arthrogryposis multiplex congenita, distal arthrogryposis, fetal akinesia deformation sequence or multiple pterygium syndrome. This sequencing was a combination of bespoke neurogenetic disease gene panels and whole exome sequencing. Only class 4 and 5 variants were reported, except for two cases where the identified variants of unknown significance (VUS) are most likely to be causative for the observed phenotype. Co-segregation studies and confirmation of variants identified by NGS were performed where possible. Functional genomics was performed as required.
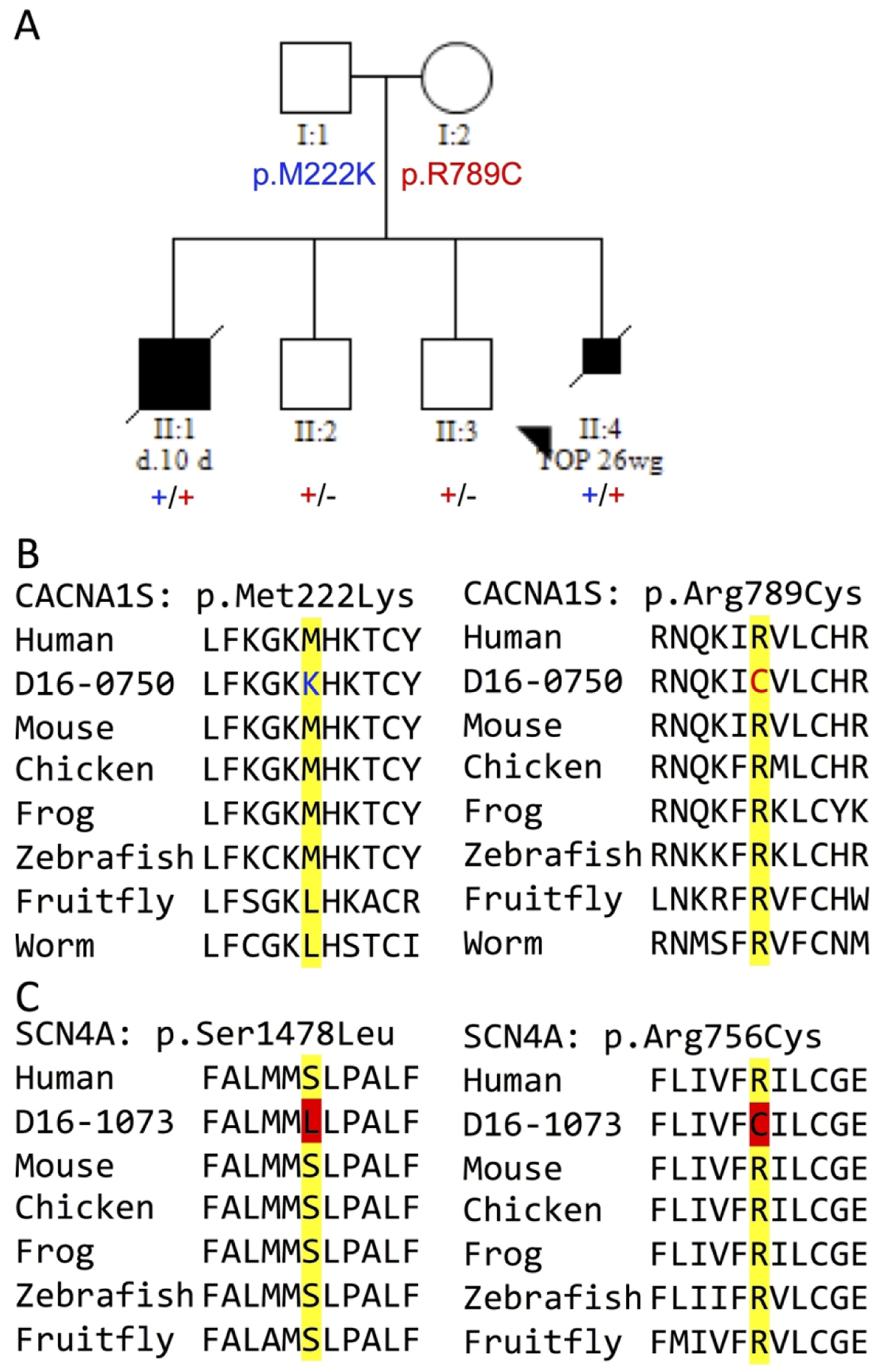
Results: Of the 190 probands, 81 received an accurate genetic diagnosis. All except two of these cases harboured class 4 and/or 5 variants based on the American College of Medical Genetics and Genomics guidelines. We identified phenotypic expansions associated with CACNA1S, CHRNB1, GMPPB and STAC3. We describe a total of 50 novel variants, including a novel missense variant in the recently identified gene for arthrogryposis with brain malformations-SMPD4.
Conclusions: Comprehensive gene panels give a diagnosis for a substantial proportion (42%) of fetal akinesia and arthrogryposis cases, even in an unselected cohort. Recently identified genes account for a relatively large proportion, 32%, of the diagnoses. Diagnostic-research collaboration was critical to the diagnosis and variant interpretation in many cases, facilitated genotype-phenotype expansions and reclassified VUS through functional genomics.
Keywords: clinical genetics; molecular genetics; neuromuscular disease.
© Author(s) (or their employer(s)) 2021. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures





References
-
- Hall JG (2009) Pena-Shokeir phenotype (fetal akinesia deformation sequence) revisited. Birth Defects Res A Clin Mol Teratol 85, 677–694 - PubMed
-
- Hall JG (2014) Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. Eur J Med Genet 57, 464–472 - PubMed
-
- Filges I, and Hall JG (2013) Failure to identify antenatal multiple congenital contractures and fetal akinesia--proposal of guidelines to improve diagnosis. Prenat Diagn 33, 61–74 - PubMed
-
- Ravenscroft G, Sollis E, Charles AK, North KN, Baynam G, and Laing NG (2011) Fetal akinesia: review of the genetics of the neuromuscular causes. J Med Genet 48, 793–801 - PubMed
-
- Todd EJ, Yau KS, Ong R, Slee J, McGillivray G, Barnett CP, Haliloglu G, Talim B, Akcoren Z, Kariminejad A, Cairns A, Clarke NF, Freckmann ML, Romero NB, Williams D, Sewry CA, Colley A, Ryan MM, Kiraly-Borri C, Sivadorai P, Allcock RJ, Beeson D, Maxwell S, Davis MR, Laing NG, and Ravenscroft G (2015) Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J Rare Dis 10, 148. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases