

Twenty years of progress in angiotensin converting enzyme 2 and its link to SARS-CoV-2 disease
- PMID: 33063823
- PMCID: PMC9055624
- DOI: 10.1042/CS20200901
Twenty years of progress in angiotensin converting enzyme 2 and its link to SARS-CoV-2 disease
Abstract
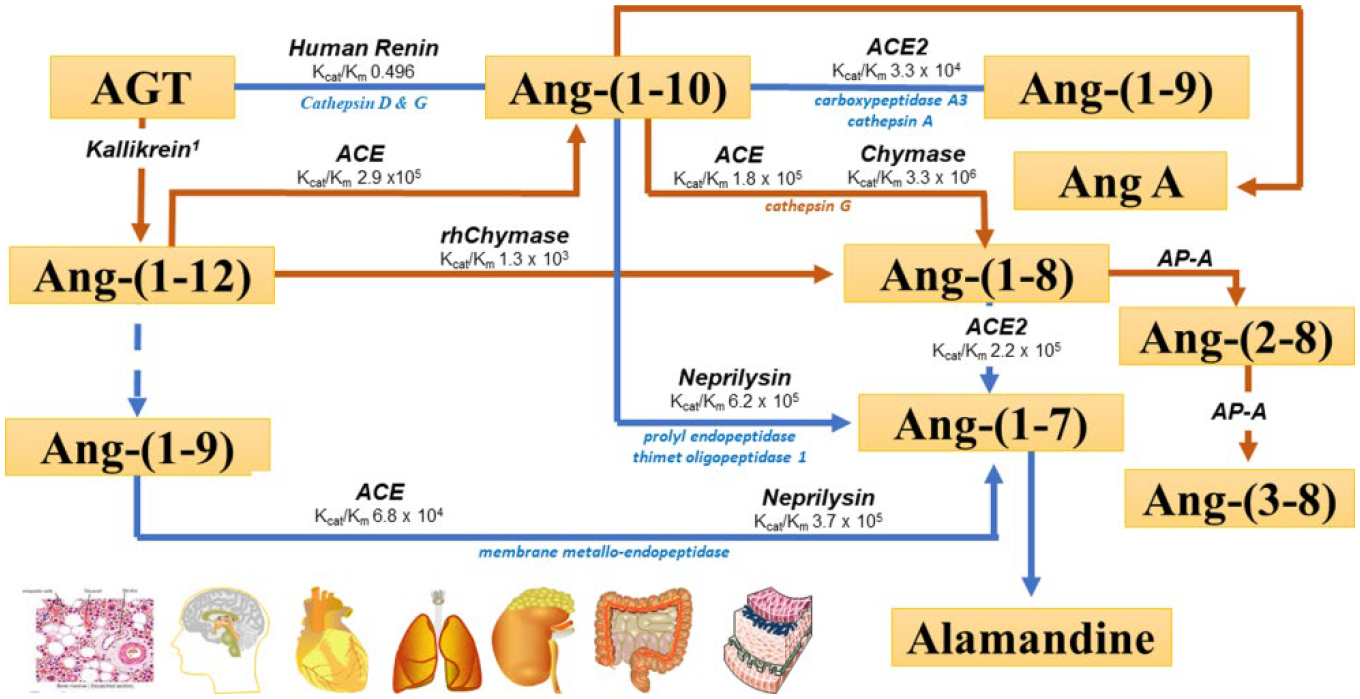
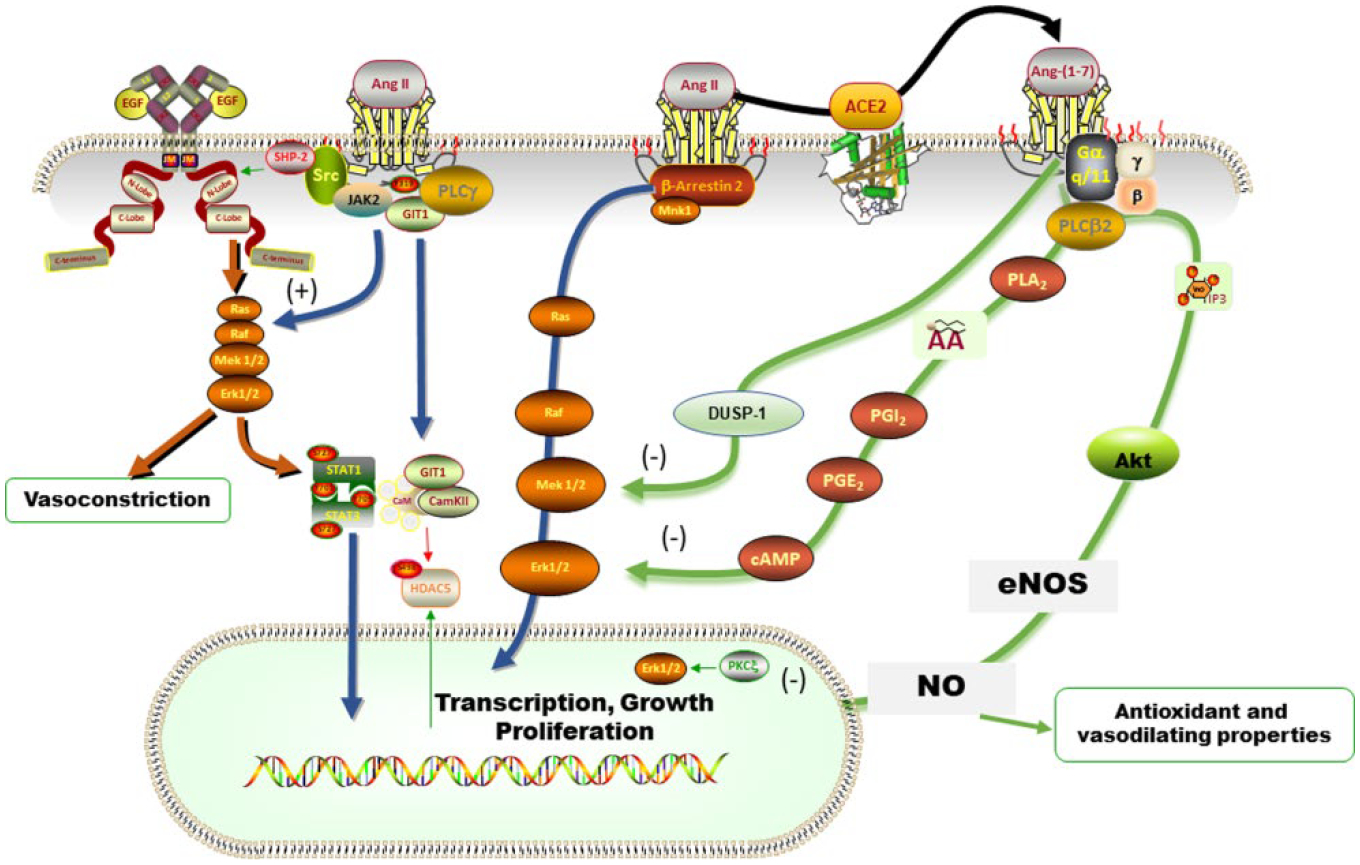
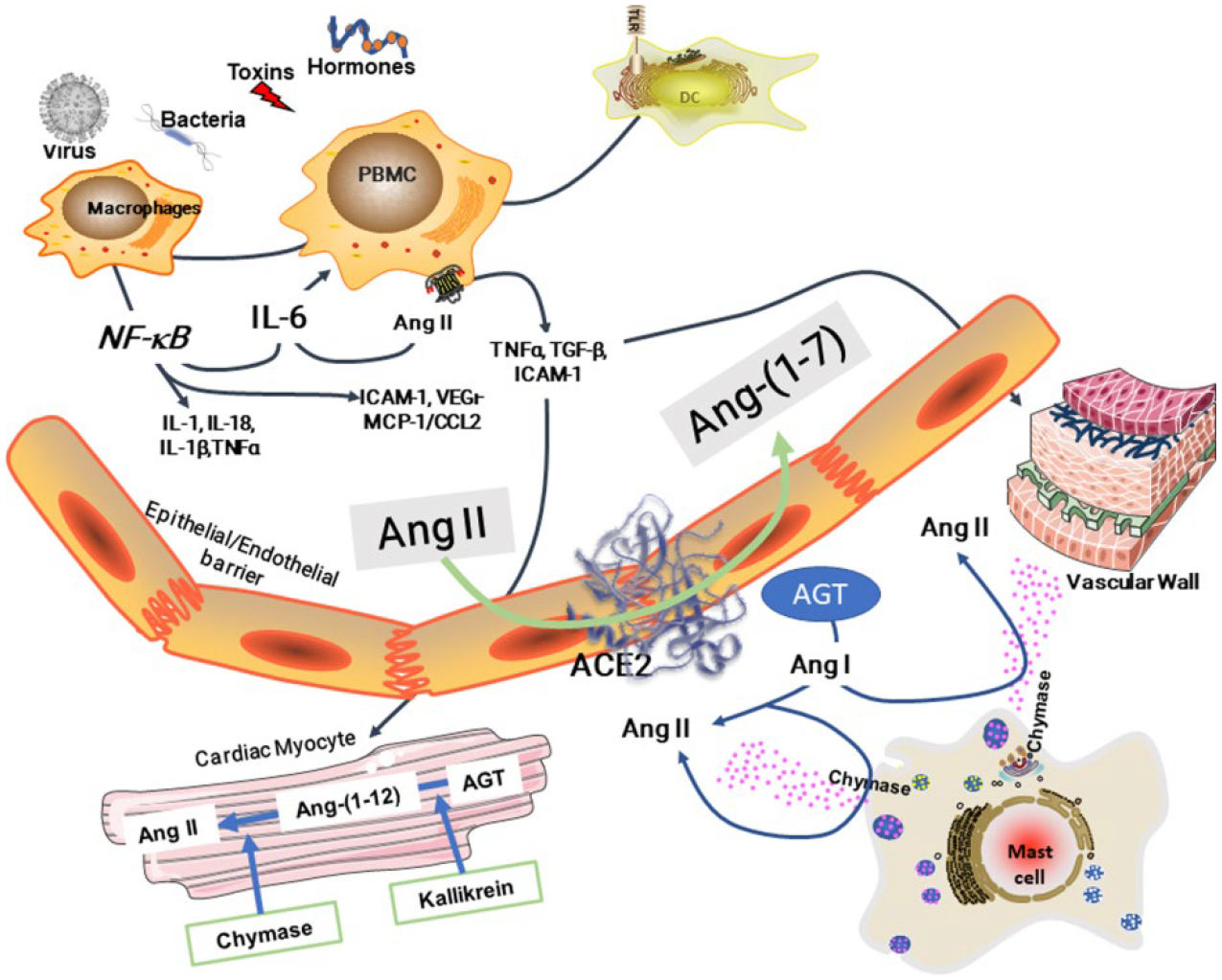
The virulence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and the aggressive nature of the disease has transformed the universal pace of research in the desperate attempt to seek effective therapies to halt the morbidity and mortality of this pandemic. The rapid sequencing of the SARS-CoV-2 virus facilitated identification of the receptor for angiotensin converting enzyme 2 (ACE2) as the high affinity binding site that allows virus endocytosis. Parallel evidence that coronavirus disease 2019 (COVID-19) disease evolution shows greater lethality in patients with antecedent cardiovascular disease, diabetes, or even obesity questioned the potential unfavorable contribution of angiotensin converting enzyme (ACE) inhibitors or angiotensin II (Ang II) receptor blockers as facilitators of adverse outcomes due to the ability of these therapies to augment the transcription of Ace2 with consequent increase in protein formation and enzymatic activity. We review, here, the specific studies that support a role of these agents in altering the expression and activity of ACE2 and underscore that the robustness of the experimental data is associated with weak clinical long-term studies of the existence of a similar regulation of tissue or plasma ACE2 in human subjects.
Keywords: angiotensin II; angiotensin-(1-7); cardiovascular disease; chymase; coronavirus; innate immunity.
© 2020 The Author(s). Published by Portland Press Limited on behalf of the Biochemical Society.
Conflict of interest statement
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Figures
Similar articles
-
Endocrine aspects of ACE2 regulation: RAAS, steroid hormones and SARS-CoV-2.J Endocrinol. 2020 Nov;247(2):R45-R62. doi: 10.1530/JOE-20-0260. J Endocrinol. 2020. PMID: 32966970 Review.
-
ACE2 and ACE: structure-based insights into mechanism, regulation and receptor recognition by SARS-CoV.Clin Sci (Lond). 2020 Nov 13;134(21):2851-2871. doi: 10.1042/CS20200899. Clin Sci (Lond). 2020. PMID: 33146371 Free PMC article. Review.
-
ACE2, the kidney and the emergence of COVID-19 two decades after ACE2 discovery.Clin Sci (Lond). 2020 Nov 13;134(21):2791-2805. doi: 10.1042/CS20200484. Clin Sci (Lond). 2020. PMID: 33135725 Review.
-
A brief review of interplay between vitamin D and angiotensin-converting enzyme 2: Implications for a potential treatment for COVID-19.Rev Med Virol. 2020 Sep;30(5):e2119. doi: 10.1002/rmv.2119. Epub 2020 Jun 25. Rev Med Virol. 2020. PMID: 32584474 Free PMC article. Review.
-
Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19.J Med Virol. 2020 Jul;92(7):726-730. doi: 10.1002/jmv.25785. Epub 2020 Apr 5. J Med Virol. 2020. PMID: 32221983 Free PMC article. Review.
Cited by
-
Combinatorial analysis of ACE and ACE2 polymorphisms reveals protection against COVID-19 worsening: A genetic association study in Brazilian patients.PLoS One. 2023 Nov 30;18(11):e0288178. doi: 10.1371/journal.pone.0288178. eCollection 2023. PLoS One. 2023. PMID: 38032879 Free PMC article.
-
The renin-angiotensin system biomolecular cascade: a 2022 update of newer insights and concepts.Kidney Int Suppl (2011). 2022 Apr;12(1):36-47. doi: 10.1016/j.kisu.2021.11.002. Epub 2022 Mar 18. Kidney Int Suppl (2011). 2022. PMID: 35529089 Free PMC article. Review.
-
ACE2, From the Kidney to SARS-CoV-2: Donald Seldin Award Lecture 2023.Hypertension. 2025 Feb;82(2):166-180. doi: 10.1161/HYPERTENSIONAHA.124.22064. Epub 2024 Dec 3. Hypertension. 2025. PMID: 39624896
-
Decrease in Angiotensin-Converting Enzyme activity but not concentration in plasma/lungs in COVID-19 patients offers clues for diagnosis/treatment.Mol Ther Methods Clin Dev. 2022 Sep 8;26:266-278. doi: 10.1016/j.omtm.2022.07.003. Epub 2022 Jul 6. Mol Ther Methods Clin Dev. 2022. PMID: 35818571 Free PMC article.
-
Plasmatic renin-angiotensin system in normotensive and hypertensive patients hospitalized with COVID-19.Biomed Pharmacother. 2022 Aug;152:113201. doi: 10.1016/j.biopha.2022.113201. Epub 2022 May 27. Biomed Pharmacother. 2022. PMID: 35661534 Free PMC article.
References
-
- Lu J, Zhou BP, Wen LX and Jiang XL (2005) Cloning of ACE-2 gene encoding the functional receptor for the SARS coronavirus and its expression in eukaryotic cells. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 19, 260–263 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous