

The design and construction of reference pangenome graphs with minigraph
- PMID: 33066802
- PMCID: PMC7568353
- DOI: 10.1186/s13059-020-02168-z
The design and construction of reference pangenome graphs with minigraph
Abstract
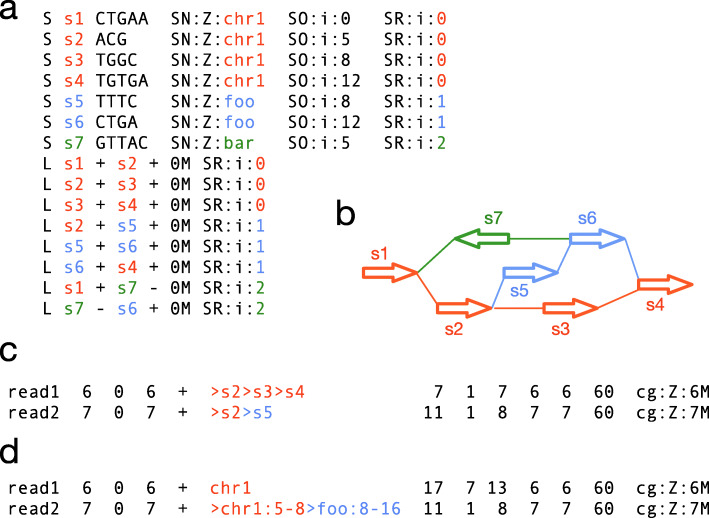
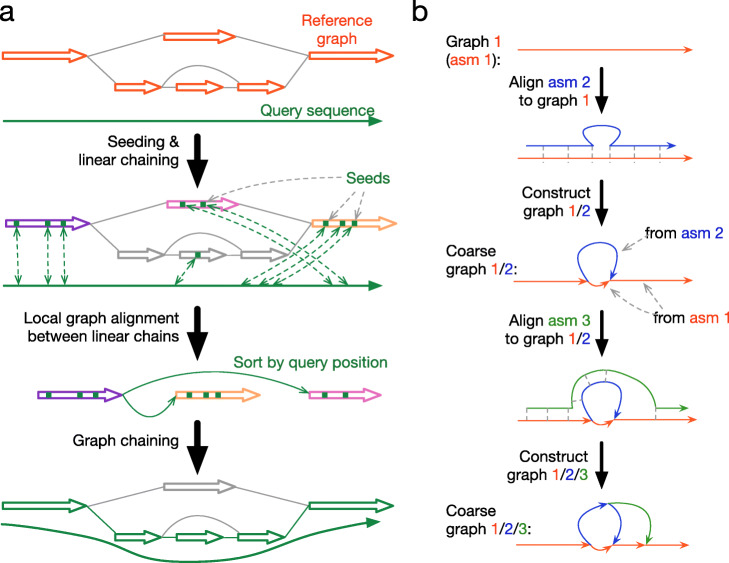
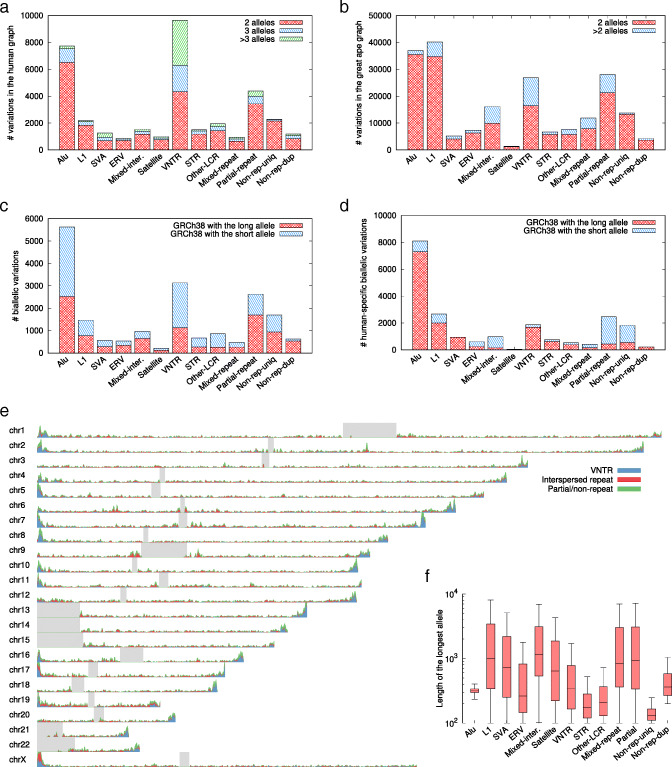
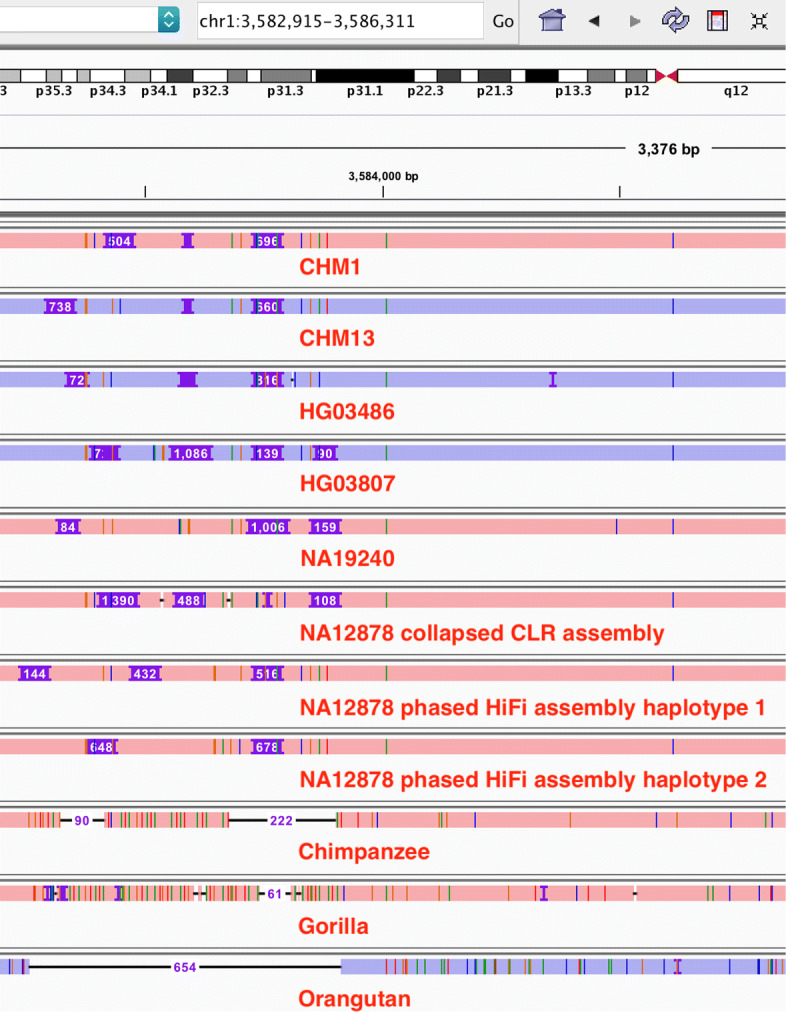
The recent advances in sequencing technologies enable the assembly of individual genomes to the quality of the reference genome. How to integrate multiple genomes from the same species and make the integrated representation accessible to biologists remains an open challenge. Here, we propose a graph-based data model and associated formats to represent multiple genomes while preserving the coordinate of the linear reference genome. We implement our ideas in the minigraph toolkit and demonstrate that we can efficiently construct a pangenome graph and compactly encode tens of thousands of structural variants missing from the current reference genome.
Keywords: Bioinformatics; Genomics; Pangenome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Pangenome graphs in infectious disease: a comprehensive genetic variation analysis of Neisseria meningitidis leveraging Oxford Nanopore long reads.Front Genet. 2023 Aug 10;14:1225248. doi: 10.3389/fgene.2023.1225248. eCollection 2023. Front Genet. 2023. PMID: 37636268 Free PMC article.
-
Comparing methods for constructing and representing human pangenome graphs.Genome Biol. 2023 Nov 30;24(1):274. doi: 10.1186/s13059-023-03098-2. Genome Biol. 2023. PMID: 38037131 Free PMC article.
-
Interactive visualization and interpretation of pangenome graphs by linear reference-based coordinate projection and annotation integration.Genome Res. 2025 Feb 14;35(2):296-310. doi: 10.1101/gr.279461.124. Genome Res. 2025. PMID: 39805704 Free PMC article.
-
Pangenome graphs and their applications in biodiversity genomics.Nat Genet. 2025 Jan;57(1):13-26. doi: 10.1038/s41588-024-02029-6. Epub 2025 Jan 8. Nat Genet. 2025. PMID: 39779953 Review.
-
The Human Pangenome Project: a global resource to map genomic diversity.Nature. 2022 Apr;604(7906):437-446. doi: 10.1038/s41586-022-04601-8. Epub 2022 Apr 20. Nature. 2022. PMID: 35444317 Free PMC article. Review.
Cited by
-
A Pangenome Approach to Detect and Genotype TE Insertion Polymorphisms.Methods Mol Biol. 2023;2607:85-94. doi: 10.1007/978-1-0716-2883-6_5. Methods Mol Biol. 2023. PMID: 36449159
-
The Need for a Human Pangenome Reference Sequence.Annu Rev Genomics Hum Genet. 2021 Aug 31;22:81-102. doi: 10.1146/annurev-genom-120120-081921. Epub 2021 Apr 30. Annu Rev Genomics Hum Genet. 2021. PMID: 33929893 Free PMC article. Review.
-
Technological Development and Advances for Constructing and Analyzing Plant Pangenomes.Genome Biol Evol. 2024 Apr 2;16(4):evae081. doi: 10.1093/gbe/evae081. Genome Biol Evol. 2024. PMID: 38669452 Free PMC article. Review.
-
Chromosome-Level Genome Assembly of the Meishan Pig and Insights into Its Domestication Mechanisms.Animals (Basel). 2025 Feb 19;15(4):603. doi: 10.3390/ani15040603. Animals (Basel). 2025. PMID: 40003085 Free PMC article.
-
Benchmarking, detection, and genotyping of structural variants in a population of whole-genome assemblies using the SVGAP pipeline.bioRxiv [Preprint]. 2025 Feb 8:2025.02.07.637096. doi: 10.1101/2025.02.07.637096. bioRxiv. 2025. PMID: 39975360 Free PMC article. Preprint.
References
-
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, Fulton RS, Kremitzki M, Magrini V, Markovic C, McGrath S, Steinberg KM, Auger K, Chow W, Collins J, Harden G, Hubbard T, Pelan S, Simpson JT, Threadgold G, Torrance J, Wood JM, Clarke L, Koren S, Boitano M, Peluso P, Li H, Chin C-S, Phillippy AM, Durbin R, Wilson RK, Flicek P, Eichler EE, Church DM. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27(5):849–64. doi: 10.1101/gr.213611.116. - DOI - PMC - PubMed
-
- Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, Graves-Lindsay TA, Munson KM, Kronenberg ZN, Vives L, Peluso P, Boitano M, Chin C-S, Korlach J, Wilson RK, Eichler EE. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017;27(5):677–85. doi: 10.1101/gr.214007.116. - DOI - PMC - PubMed
-
- Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37(10):1155–62. doi: 10.1038/s41587-019-0217-9. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
