

OpenPepXL: An Open-Source Tool for Sensitive Identification of Cross-Linked Peptides in XL-MS
- PMID: 33067342
- PMCID: PMC7710140
- DOI: 10.1074/mcp.TIR120.002186
OpenPepXL: An Open-Source Tool for Sensitive Identification of Cross-Linked Peptides in XL-MS
Abstract
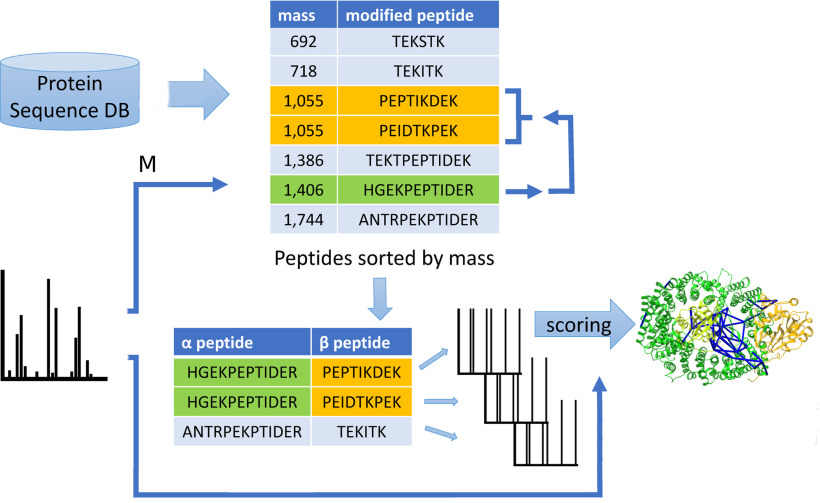
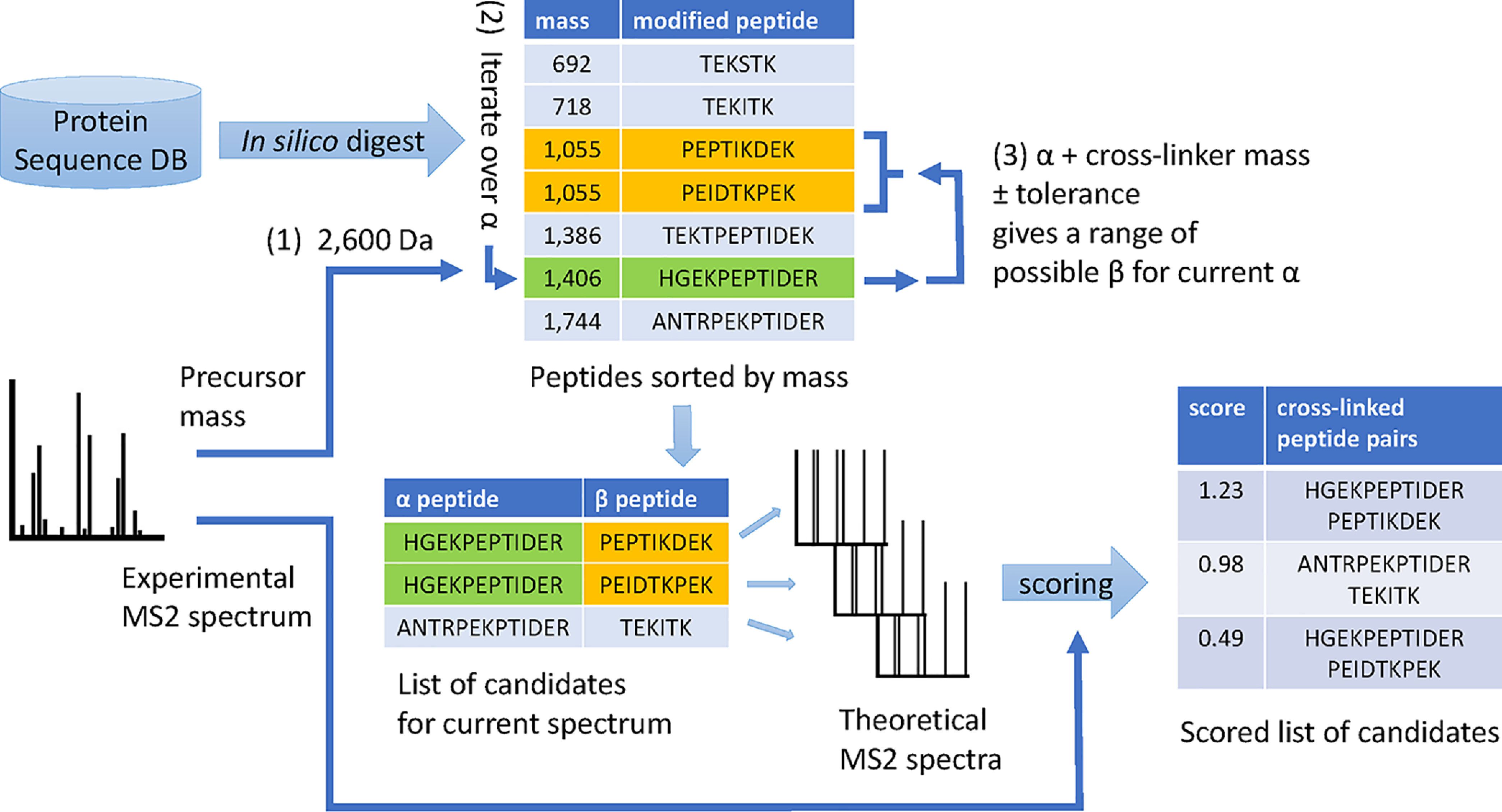
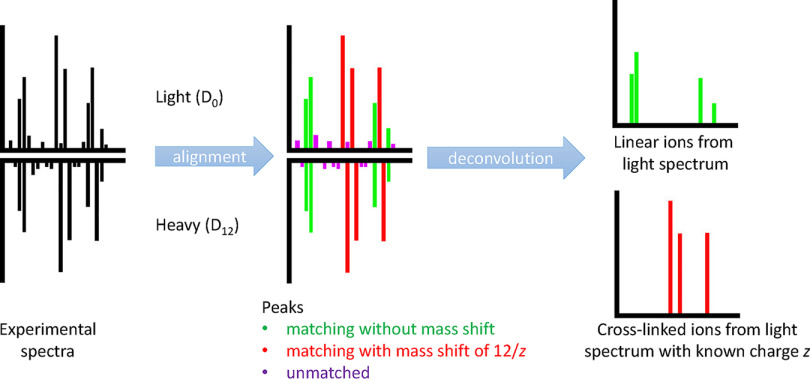
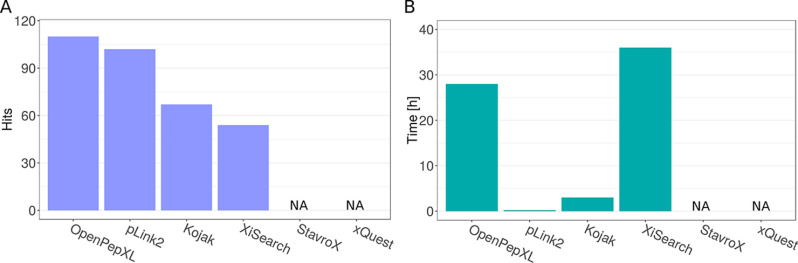
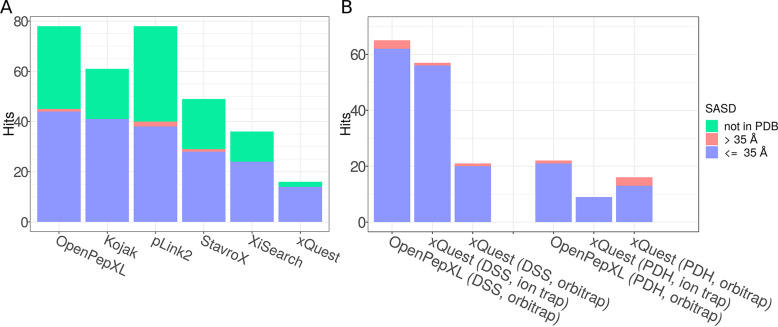
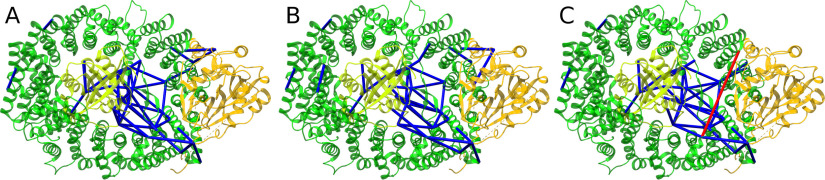
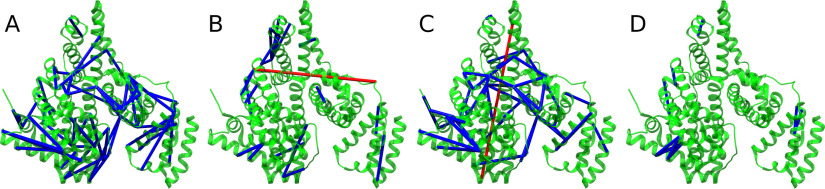
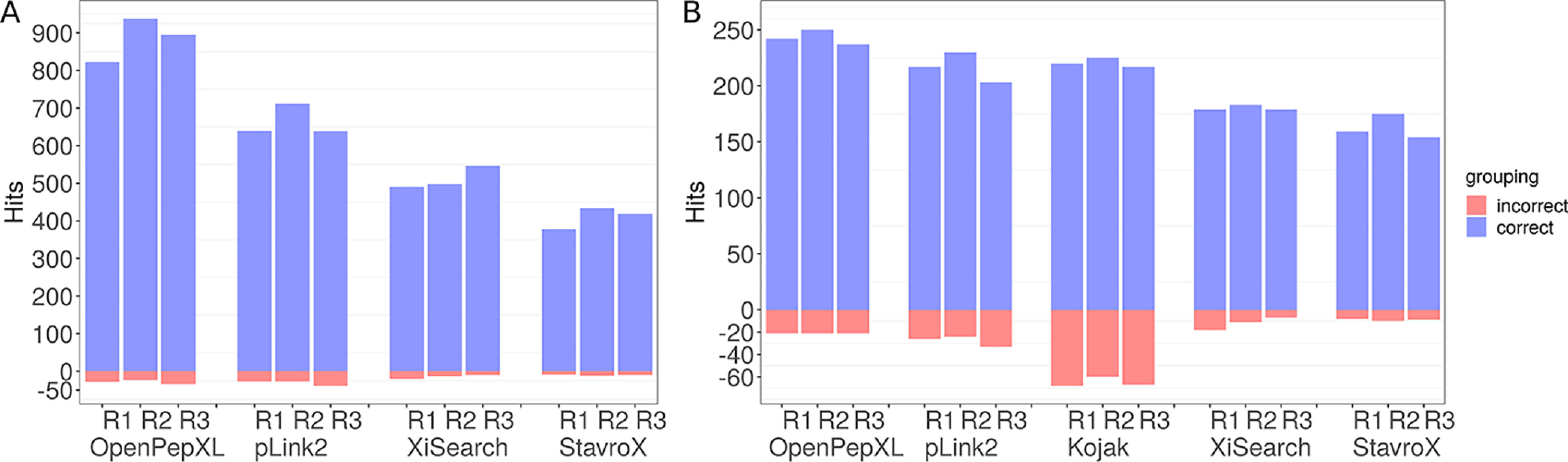
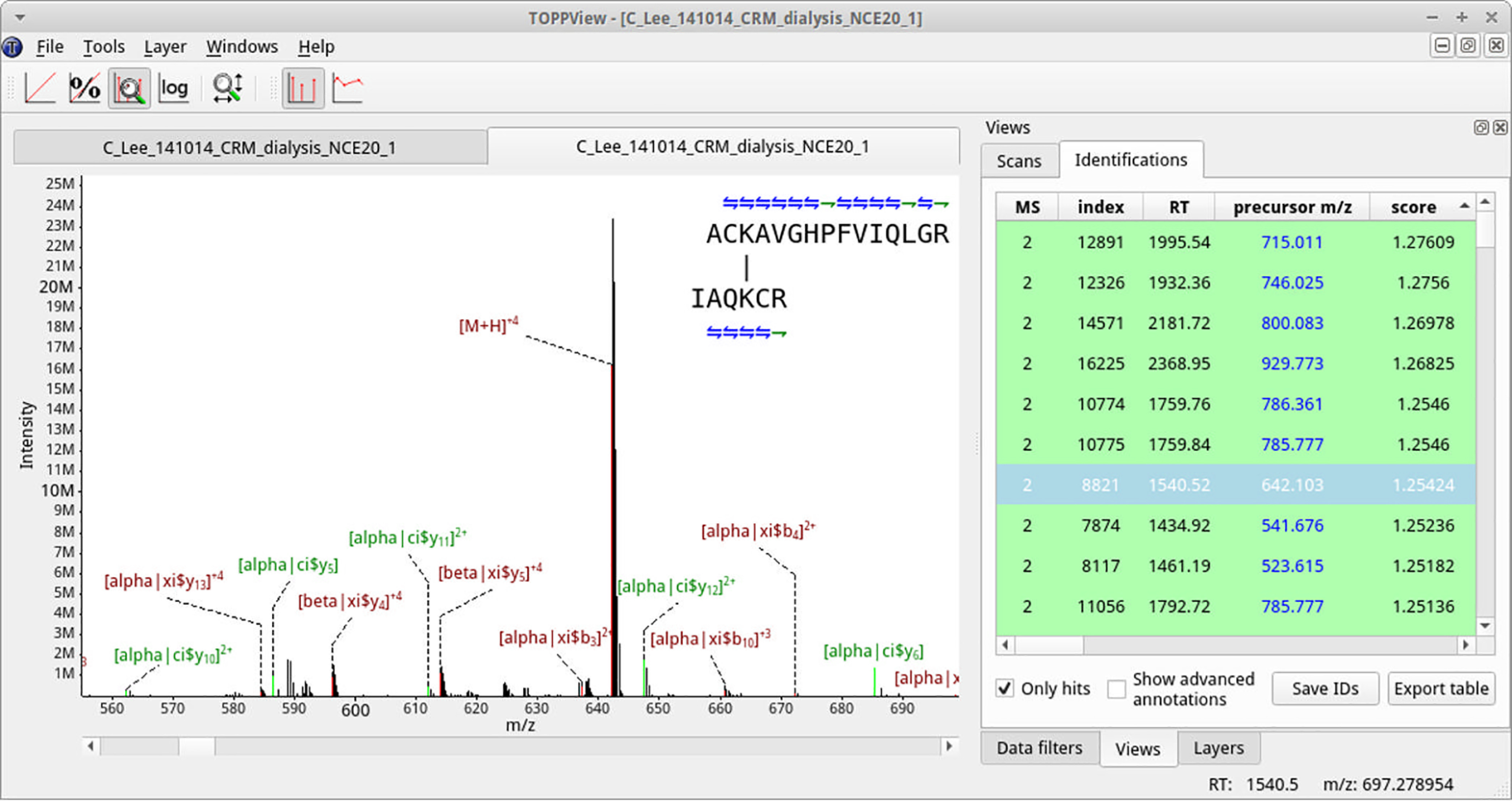
Cross-linking MS (XL-MS) has been recognized as an effective source of information about protein structures and interactions. In contrast to regular peptide identification, XL-MS has to deal with a quadratic search space, where peptides from every protein could potentially be cross-linked to any other protein. To cope with this search space, most tools apply different heuristics for search space reduction. We introduce a new open-source XL-MS database search algorithm, OpenPepXL, which offers increased sensitivity compared with other tools. OpenPepXL searches the full search space of an XL-MS experiment without using heuristics to reduce it. Because of efficient data structures and built-in parallelization OpenPepXL achieves excellent runtimes and can also be deployed on large compute clusters and cloud services while maintaining a slim memory footprint. We compared OpenPepXL to several other commonly used tools for identification of noncleavable labeled and label-free cross-linkers on a diverse set of XL-MS experiments. In our first comparison, we used a data set from a fraction of a cell lysate with a protein database of 128 targets and 128 decoys. At 5% FDR, OpenPepXL finds from 7% to over 50% more unique residue pairs (URPs) than other tools. On data sets with available high-resolution structures for cross-link validation OpenPepXL reports from 7% to over 40% more structurally validated URPs than other tools. Additionally, we used a synthetic peptide data set that allows objective validation of cross-links without relying on structural information and found that OpenPepXL reports at least 12% more validated URPs than other tools. It has been built as part of the OpenMS suite of tools and supports Windows, macOS, and Linux operating systems. OpenPepXL also supports the MzIdentML 1.2 format for XL-MS identification results. It is freely available under a three-clause BSD license at https://openms.org/openpepxl.
Keywords: Protein cross-linking; XL-MS; crosslinking; protein structure; structural biology; tandem mass spectrometry.
© 2020 Netz et al.
Conflict of interest statement
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Liu F., and Heck A. J. (2015) Interrogating the architecture of protein assemblies and protein interaction networks by cross-linking mass spectrometry. Curr. Opin. Struct. Biol. 35, 100–108 - PubMed
-
- Leitner A., Faini M., Stengel F., and Aebersold R. (2016) Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci. 41, 20–32 - PubMed
-
- O'Reilly F. J., and Rappsilber J. (2018) Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat. Struct. Mol. Biol. 25, 1000–1008 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
